
Noble-metal free catalysts for electrochemical water splitting: Recent progress and perspectives on application
-
摘要:
利用可再生电能分解水制取氢气是理想的绿色制氢方式,是氢能技术发展的基础,可有效缓解能源消耗对环境的污染,推动我国碳达峰碳中和目标的实现. 然而,电解水制氢过程需要消耗大量电能,且受到高昂的催化剂成本的限制,电解水制氢技术的规模化、可持续发展存在阻力. 因此,设计制备低成本、高性能的过渡金属基催化剂,以及选择热力学上更易发生的氧化反应替代高能垒的析氧反应,是提高制氢效率的主要策略. 本文系统地总结了非贵过渡金属基催化剂在电解水制氢中的应用,以及阳极替代反应在降低制氢能耗方面的最新研究进展. 此外,针对降低电解水制氢技术能耗,推进大规模、绿色节能的工业电解水制氢发展面临的挑战和新的机遇进行了展望.
-
关键词:
- 电催化分解水 /
- 析氢反应 /
- 析氧反应 /
- 非贵过渡金属基电催化剂 /
- 阳极替代反应
Abstract:Advanced water electrolysis powered by renewable energy is the most ideal and environmentally friendly approach for hydrogen production, serving as a technological foundation for large-scale hydrogen energy applications. This process can significantly reduce environmental pollution from energy consumption and support China’s carbon neutrality goals. However, the high energy demands and costs of noble metals pose challenges to scaling up hydrogen production from water electrolysis. To enhance efficiency, developing low-cost yet highly efficient noble metal-free electrocatalysts for the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) is crucial. Understanding the mechanisms behind HER and OER helps identify factors affecting electrocatalyst efficiency and design strategies to improve performance. Moreover, replacing the energy-intensive OER with more energy-efficient reactions offers another promising way to promote hydrogen production. This review summarizes recent advancements in nonprecious transition metal-based electrocatalysts for water electrolysis. Compared to noble metal-based electrocatalysts, nonprecious transition metal-based electrocatalysts like Fe, Co, and Ni-based oxides, (oxy) hydroxides, chalcogenides, and their derivates offer abundant reserves, lower costs, and adjustable catalytic properties, making them viable alternatives for large-scale water splitting. Understanding how these materials catalyze HER and the OER in different electrolytes is key to designing strategies, such as element doping, hetero-structuring, lattice defect construction, carbon composite coupling, and surface reconstruction, to reduce energy costs of electrochemical water splitting. The mechanisms behind these strategies for enhancing water electrolysis are explained through the thermodynamics of absorbed intermediates and the reaction kinetics. Beyond reducing overpotentials, another strategy involves replacing OER with the anodic oxidation reaction of organic molecules, effectively lowering the overall voltage. This review highlights recent progress and strategies for designing efficient electrocatalysts for the anodic oxidation of diverse organics, including urea, amine, hydrazine, alcohol, aldehyde, and sulfates, in substitution of water molecules. This review also addresses the gap between lab-scale research and industry-scale application of hydrogen production. It considers research on water splitting mechanisms, catalyst development, and OER-substituting electrooxidation reactions alongside electrolyzer design, synthesis costs, working conditions, and evaluation criteria. It also compares recent advancements in state-of-the-art water electrolysis technologies and summarizes their application prospects in hydrogen production. The review aims to provide theoretical guidance for designing and synthesizing advanced transition-metal-based electrocatalysts for HER, OER, and substitution anodic reactions for energy-efficient hydrogen production while also shedding light on opportunities for energy-efficient hybrid water-splitting applications.
-
随着人类社会的快速发展,能源消耗持续增加. 化石燃料作为主要能量来源之一日益短缺,并且其燃烧产物排放引起的气候变暖和环境污染问题日益严重. 因此,大力发展可再生能源,构建清洁低碳能源体系,推进新一轮的能源革命,是实现我国双碳目标的重要举措[1−3]. 氢的能量密度高,燃烧或氧化后的唯一产物是环境友好的水,因此氢成为未来可再生能源转化、存储和利用的理想能源载体[4]. 在我国《氢能产业发展中长期规划(2021—2035年)》中明确指出氢能产业是战略性新兴产业和未来产业重点发展方向,而实现大规模绿色制氢是氢能产业发展的重要环节[5]. 相比于石油裂解、甲烷蒸汽重整及水煤气转化的制氢方式,电解水制氢能够利用可再生电能驱动水分解制备高纯度的氢气,该过程中不存在碳排放,因此被认为是最理想的制氢方式. 水分解反应是一个吉布斯自由能(ΔG = 237.1 kJ·mol−1)增加的非自发反应,因此理论上需要施加1.23 V的热力学平衡电位以驱动电解水反应[6]. 然而,在实际的电解水反应中,阴极析氢和阳极析氧的动力学过程缓慢,需要额外施加过电位以克服动力学能垒.
为降低阴/阳极过电位,减少电解水制氢的能耗,设计制备高效的催化材料是至关重要的策略. 现阶段,催化阳极析氧反应(Oxygen evolution reaction, OER)的商用材料主要是Ir、Ru贵金属及其氧化物;催化阴极析氢反应(Hydrogen evolution reaction, HER)的主要是Pt基材料. 尽管上述电极材料具有优异的催化活性,但其高昂的成本和高负载量限制了电解水制氢经济效益的提高. 非贵过渡金属基电催化剂具有地壳储量丰富,价格低廉等显著优势,其较好的催化性能展现出取代贵金属基电催化剂的潜力. 因此,分别基于阴/阳极半反应针对性地设计高效、低成本、性能稳定的非贵过渡金属基电催化剂,是实现工业电解水制氢经济化运行的关键.
除了反应过电位以外,电解水反应过高的热力学平衡电位是造成电解槽耗能多的主要原因. 相比于HER,OER涉及更为复杂的电子转移过程,需要更多的电能来克服阳极反应能垒. 因此,选择热力学平衡电位更低,更容易发生的氧化反应替代OER辅助制氢,能够进一步降低能耗,推动大规模工业节能制氢的发展. 降低催化剂的成本和电解槽的能耗是推动电解水制氢工业规模化、可持续发展的重中之重. 本文首先总结非贵过渡金属基催化剂在HER和OER中的应用,归纳提升材料催化性能的关键策略,为工业电解水制氢技术提供了材料支持. 进而介绍阳极替代反应辅助制氢的研究进展和工程应用前景. 针对目前的研究现状,本文讨论了大规模制备高效非贵过渡金属基催化剂和电解水制氢技术的最新发展方向,为推动节能电解水制氢工业发展提供了参考.
1. 电解水制氢的基本原理
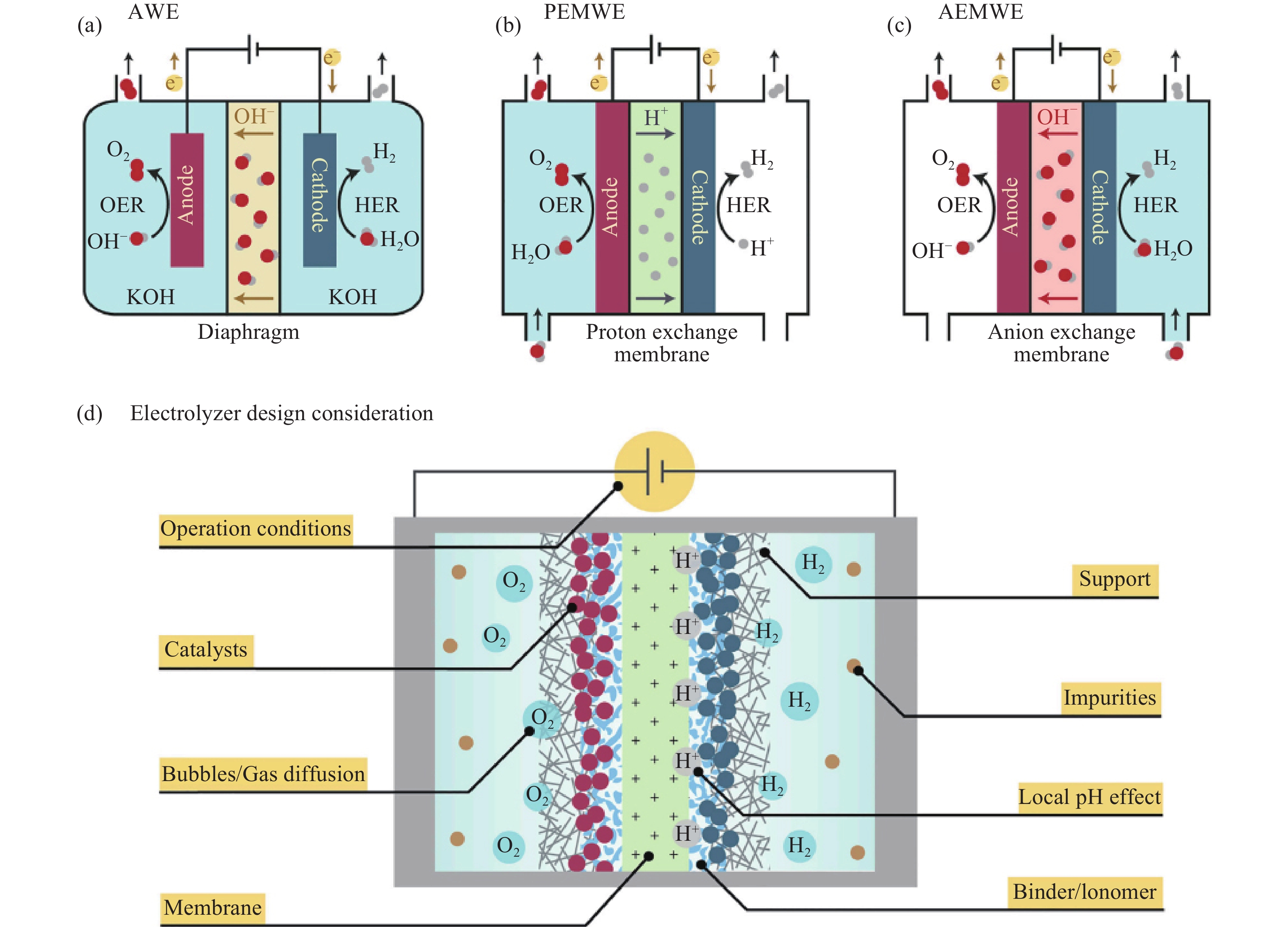
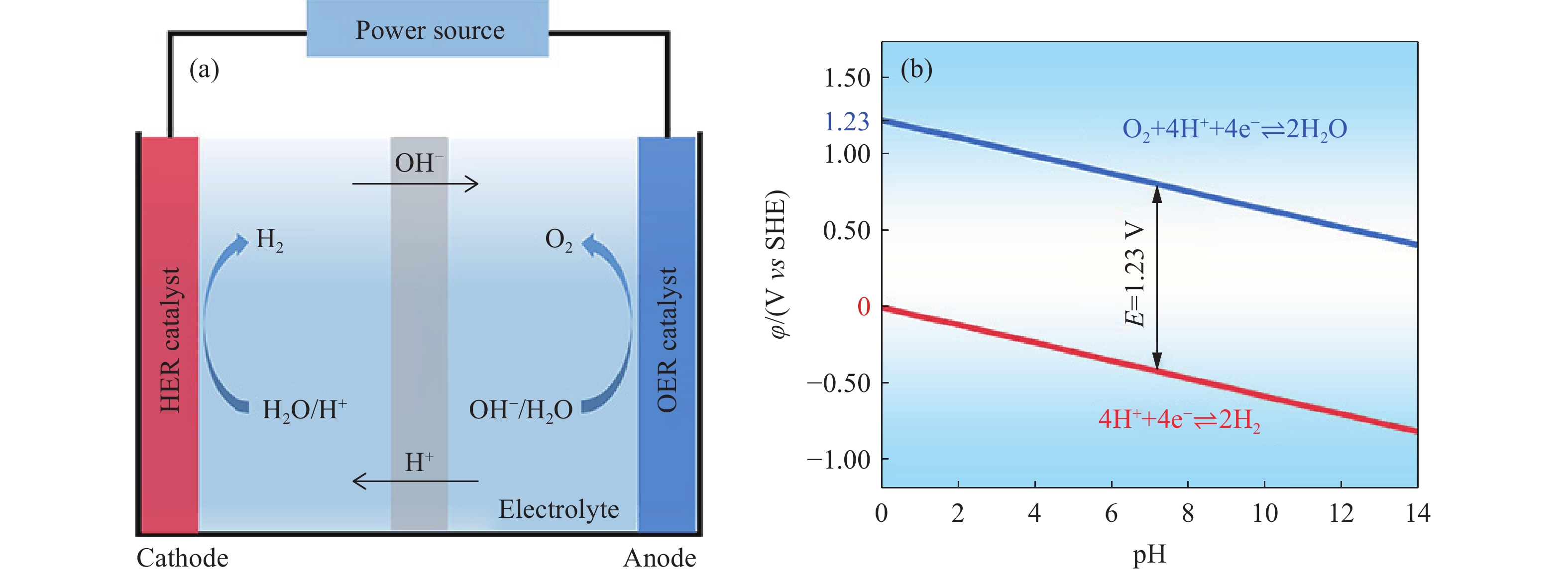
电解水反应的热力学平衡电位为1.23 V,由两个半反应组成:在阴极发生析氢反应和在阳极发生的析氧反应. 如图1 (a) 所示,电解水反应发生在由电解液、阴极和阳极三部分组成的电解槽中,通常在阴、阳极上分别负载HER和OER催化剂以加速水分解.
![]() 图 1 (a) 电解槽示意图; (b) HER和OER的电极电位随pH变化的Pourbaix图Figure 1. (a) Scheme of a water electrolyzer; (b) Pourbaix plot of electrode potential vs pH for the HER and OER
图 1 (a) 电解槽示意图; (b) HER和OER的电极电位随pH变化的Pourbaix图Figure 1. (a) Scheme of a water electrolyzer; (b) Pourbaix plot of electrode potential vs pH for the HER and OER在不同的pH条件下,电解液中的H+ 和OH− 浓度不同,对应的电解水反应中阴、阳极的反应物也有所不同[7]. 在酸性电解质溶液中:
$$ \mathrm{阴极:4H}^{ \mathrm+} \mathrm{+4e}^{ -}\to \mathrm{2H}_{ \mathrm{2}} $$ (1) $$ \mathrm{阳极:2H}_{ \mathrm{2}} \mathrm{O\to4H}^{ \mathrm+} \mathrm{+4e}^{ -} \mathrm{+O}_{ \mathrm{2}} $$ (2) 在中性和碱性电解质溶液中:
$$ \mathrm{阴极:4H}_{ \mathrm{2}} \mathrm{O+4e}^{ -} \to\mathrm{4OH}^{ -} \mathrm{+2H}_{ \mathrm{2}} $$ (3) $$ \mathrm{阳极:4OH}^{ -} \to\mathrm{2H}_{ \mathrm{2}} \mathrm{O+4e}^{ -} \mathrm{+O}_{ \mathrm{2}} $$ (4) 根据能斯特方程,以标准氢电极(Standard hydrogen electrode, SHE)作为参比电极,HER和OER的电极电位依赖于pH的变化,如图1 (b) 所示. 与SHE相比,可逆氢电极(Reversible hydrogen electrode, RHE)考虑了pH对电极电势的影响(φRHE = φSHE + 0.059pH),以此为参照能够方便地比较电催化剂在不同pH条件下的电极电势,因此电解水的研究中常以RHE作为参比电极.
由于反应能垒的存在,需在电极上施加足够的过电位(η)以降低反应的活化能能垒,才能获得可观的电解水反应速率. 因此,电解水反应的实际电压(E)如式 (5) 所示:
$$ \mathit{E} \mathrm{=1.23V}+ \mathit{\eta } _{ \mathrm{cathode}} + \mathit{\eta } _{ \mathrm{anode}} + \mathit{I} \mathrm{\times } \mathit{R} _{ \mathrm{cell}} $$ (5) 式中,ηcathode表示阴极过电位,ηanode表示阳极过电位,I表示电流,Rcell表示电解槽电阻. 开发高效的HER和OER催化剂是降低反应过电位,从而减少电能消耗最有效的方式. 在电催化分解水的研究中,为方便比较不同催化剂之间的性能,通常选用0.5~2、10、100和
1000 mA·cm−2电流密度(j)所对应的起峰过电位(ηonset)、η10、η100和η1000 作为评价催化剂性能的标准.1.1 HER机理
HER过程的实质是H+在电极/溶液界面处被还原成H2. 反应路径分为Volmer–Heyrovsky和Volmer–Tafel两种路径[8]. 在酸性电解质溶液中,析氢反应的历程式(6)~(9)所示[9]:
$$ \mathrm{Volmer步骤:H}^{ \mathrm+} \mathrm{+e}^{ -} \mathrm{+^*\to ^*H} $$ (6) $$ \mathrm{Heyrovsky步骤:H}^{ \mathrm+} \mathrm{+e}^{ -} \mathrm{+^*H\to H}_{ \mathrm{2}} \mathrm{+^*} $$ (7) $$ \mathrm{Tafel步骤:^*H+^*H\to H}_{ \mathrm{2}} \mathrm{+2^*} $$ (8) 式中,*代表电极表面的电催化活性位点,*H表示吸附的氢原子. 而在碱性和中性电解质溶液中,由于H+ 浓度极低,H2O会优先在电极表面发生解离,从而产生大量的H+ 形成 *H,进而在催化剂表面发生电子转移,释放H2.
在实际电化学反应中,Tafel斜率能够表示反应动力学步骤的快慢,可以用来推测反应的速控步骤. 在HER中,当Tafel斜率接近120 mV·dec−1时,Volmer步骤是限速步骤;当Tafel斜率接近40 mV·dec−1时,Heyrovsky步骤是限速步骤;当Tafel斜率接近30 mV·dec−1时,Tafel步骤是限速步骤.
1.2 OER机理
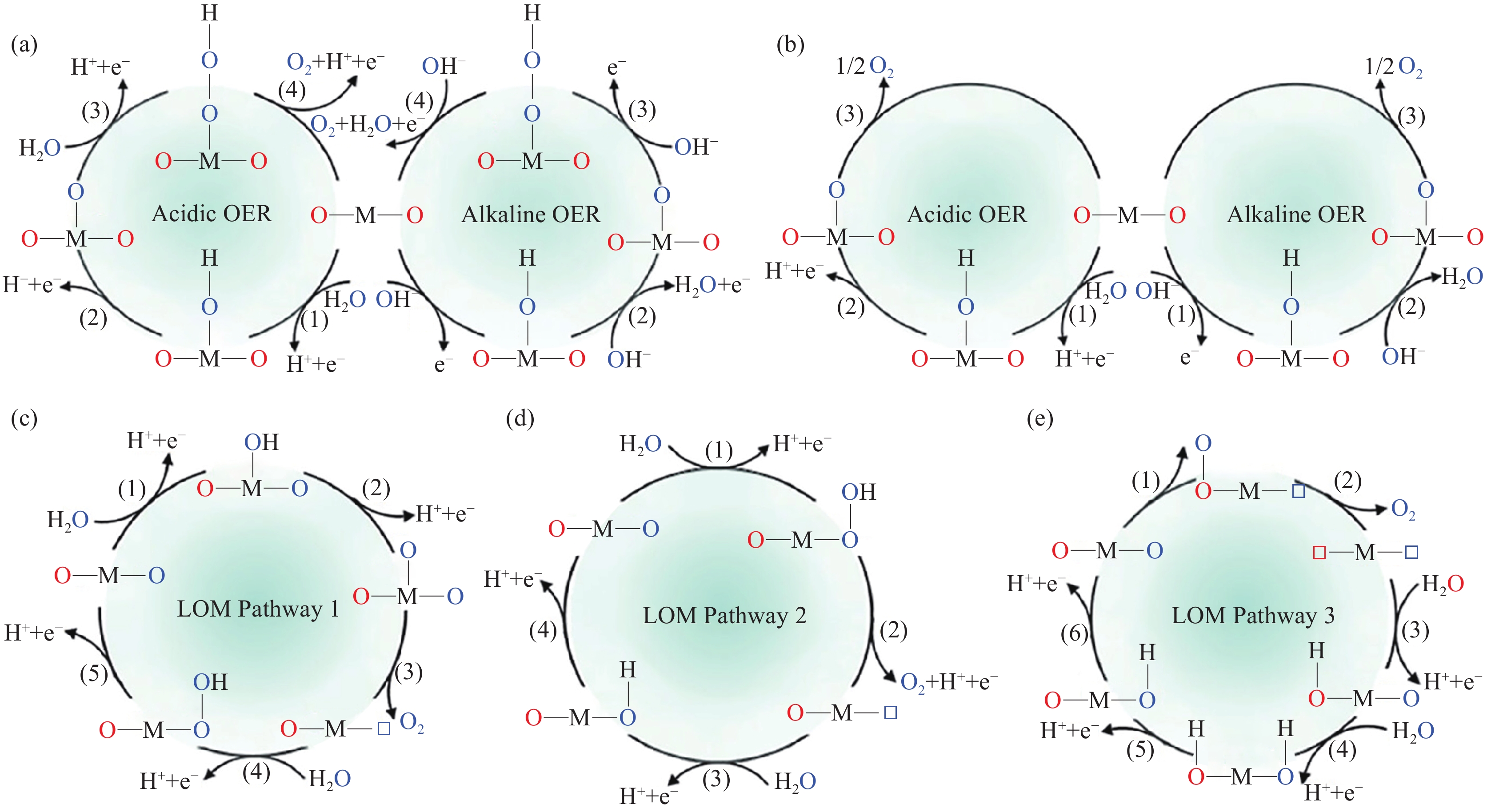
OER的反应机制主要包括传统的吸附质演化机制(Adsorbent evolution mechanism, AEM)和晶格氧机制(Lattice oxygen mechanism, LOM)两种. 对于AEM反应机制,在酸性电解质溶液中OER生成吸附羟基*OH和吸附氧原子*O的步骤如式(9)和(10)所示[10],对应于图2(a)~(b)中的步骤(1)~(2). 由 *O生成O2通常有两种方式[11]:一种是 *O与H2O结合,脱去H+和e−形成 *OOH中间物种,*OOH再进一步脱去H+释放O2,对应于图2(a)中的步骤(3)~(4)及式(11)~(12);另一种是两个近邻的 *O直接结合生成吸附O2,对应于图2(b)中的步骤(3)及式(13).
![]() 图 2 (a)~(b) OER过程中的AEM机制; (c)~(e) OER过程中的LOM机制[14]. 其中,M代表金属元素,蓝色O代表电解液中的羟基氧,红色O代表晶格氧,方框代表氧空位Figure 2. (a)–(b) AEM mechanism scheme of the OER; (c)–(e) LOM mechanism scheme of the OER[14]. M represents the metal element, blue O, red O, and box represents the oxygen in hydroxyl, the lattice oxygen, and the box represents the oxygen vacancy, respectively
图 2 (a)~(b) OER过程中的AEM机制; (c)~(e) OER过程中的LOM机制[14]. 其中,M代表金属元素,蓝色O代表电解液中的羟基氧,红色O代表晶格氧,方框代表氧空位Figure 2. (a)–(b) AEM mechanism scheme of the OER; (c)–(e) LOM mechanism scheme of the OER[14]. M represents the metal element, blue O, red O, and box represents the oxygen in hydroxyl, the lattice oxygen, and the box represents the oxygen vacancy, respectively$$ \mathrm{^*+H}_{ \mathrm{2}} \mathrm{O\to ^*OH+H}^{ \mathrm+} \mathrm{+e}^{ -} $$ (9) $$ \mathrm{^*OH\to ^*O+H}^{ \mathrm+} \mathrm{+e}^{ -} $$ (10) $$ \mathrm{^*O+H}_{ \mathrm{2}} \mathrm{O\to ^*OOH+H}^{ \mathrm+} \mathrm{+e}^{ -} $$ (11) $$ \mathrm{^*OOH\to O}_{ \mathrm{2}} \mathrm{+^*+H}^{ \mathrm+} \mathrm{+e}^{ -} $$ (12) $$ \mathrm{^*O+^*O\to O}_{ \mathrm{2}} \mathrm{+2^*} $$ (13) 在碱性和中性环境中,OH−直接吸附在*上,连续脱去e− 形成*OH、*O、*OOH中间物种,最后释放O2(图2(a)~(b)). OER涉及四电子传递过程,当Tafel斜率分别接近120、40、24和15 mV·dec−1时,OER过程的限速步骤分别为形成吸附羟基*OH的步骤、形成吸附氧*O的步骤、形成中间产物*OOH的步骤和释放O2的步骤.
随着原位表征技术的发展,在反应过程中特别是高过电位的条件下观察到了催化剂表面发生了动态结构的变化[12−13],传统的AEM机制无法解释该实验现象,由此研究人员提出了新的LOM机制. 其中,材料中的晶格氧在酸性条件下参与OER的反应式如下[14]:
$$ \mathrm{^*+H}_{ \mathrm{2}} \mathrm{O\to ^*OH+H}^{ \mathrm+} \mathrm{+e}^{ -} $$ (14) $$ \mathrm{^*OH\to ^*O+H}^{ \mathrm+} \mathrm{+e}^{ -} $$ (15) $$ \mathrm{^*O+O}_{ \mathrm{L}} \mathrm{\to O}_{ \mathrm{2}} \mathrm{+V}_{ \mathrm{O}} $$ (16) $$ \mathrm{或^*OH+O}_{ \mathrm{L}} \mathrm{\to O}_{ \mathrm{2}} \mathrm{+H}^{ \mathrm+} \mathrm{+e}^{ -} \mathrm{+V}_{ \mathrm{O}} $$ (17) $$ \mathrm{V}_{ \mathrm{O}} \mathrm{+H}_{ \mathrm{2}} \mathrm{O\to ^*OH+H}^{ \mathrm+} \mathrm{+e}^{ -} $$ (18) $$ \mathrm{^*H\to ^*+H}^{ \mathrm+} \mathrm{+e}^{ -} $$ (19) LOM机制的第一步与AEM机制类似,对应于图2(c)~(e)中的步骤(1). 由于*OH中间体与晶格氧原子(OL)结合,生成O2和氧空位(VO)的方式不同,LOM机制分为三种反应路径:第一种是*OH脱去H+形成*O,*O与OL结合释放O2并形成VO,对应于图2(c)的步骤(2)~(3)及式(15)~(16);第二种是吸附在O位点的*OH直接与OL结合脱去H+形成VO,对应于图2(d)的步骤(2)及式(17);第三种是两个OL结合释放氧气,形成两个VO,对应于图2(e)的步骤(2). 最后,H2O中的O原子填入VO,脱去H+释放活性位点*,对应于图2(c)~(e)中的后续步骤.
2. 提高电催化分解水效率的策略
电催化分解水反应是一个耗能反应[6],需要足够的过电位克服阴极/阳极反应的活化能能垒. 相比于HER,OER具有更高的热力学平衡电位,限制了电催化分解水效率的提升[11]. 因此,本文总结了两种提高电催化分解水效率的策略:一是开发经济高效的非贵过渡金属基HER和OER催化剂,另一个则是选择热力学平衡电位更低的阳极替代反应辅助电催化分解水制氢.
2.1 基于非贵过渡金属基催化剂提高电解水效率的策略
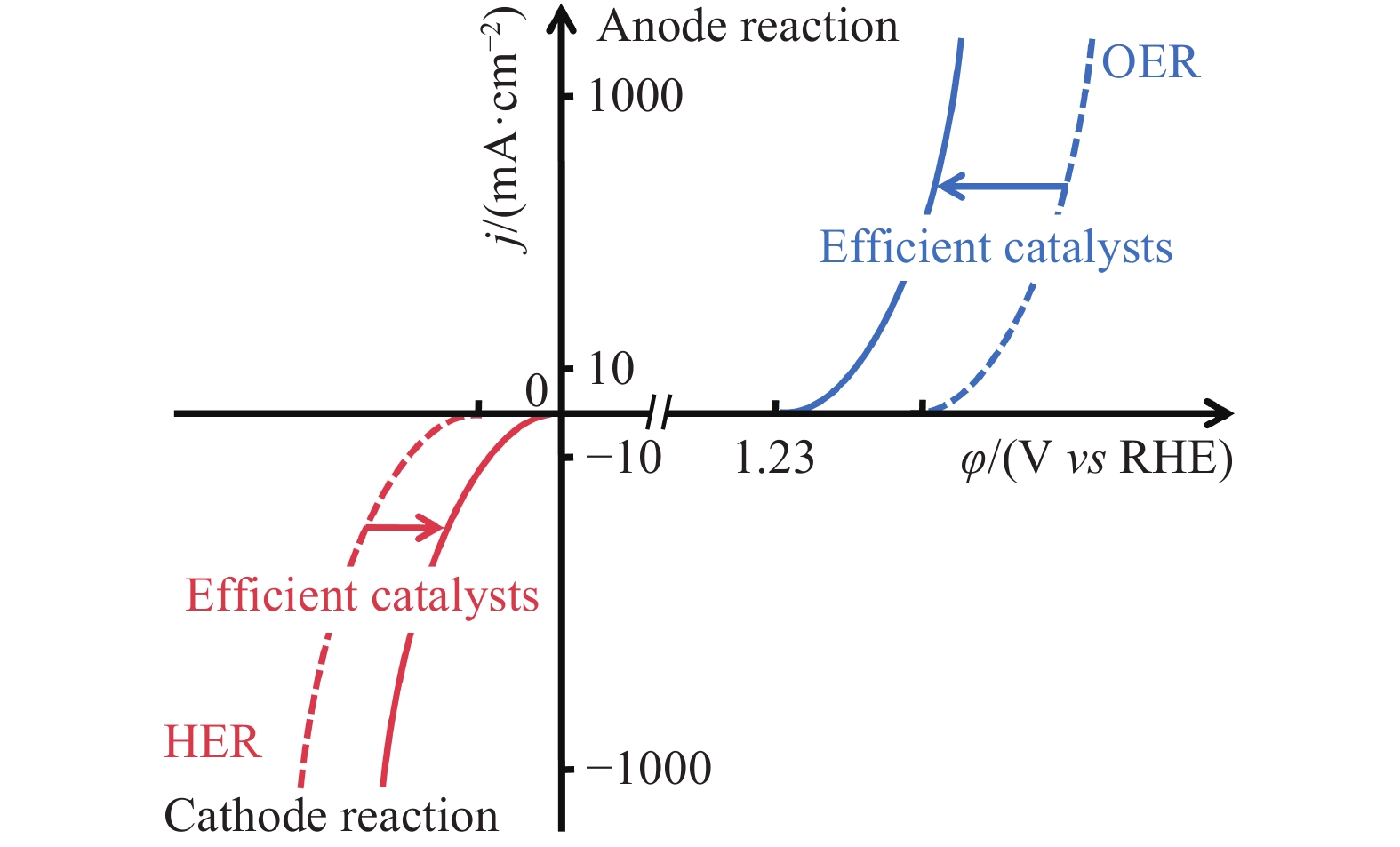
图3中实线表示催化剂的理想性能,虚线表示实际条件下催化剂的性能. 其中,HER热力学理论开始分解电压为0 V vs RHE,OER热力学理论开始分解电压为1.23 V vs RHE,由于反应过电位和串联电阻的存在,实际开始的分解电压明显高于理论分解电压, 要达到工业要求的安培级反应电流密度则需要更高的分解电压[15]. 即图3中制备高活性的催化剂促使阴极极化曲线向右移动,阳极极化曲线向左移动,降低HER和OER的反应过电位,从而有效提高电解水效率.
![]() 图 3 理想条件下和实际应用中,HER(红色实线)和OER(蓝色实线)的线性扫描伏安(Linear sweep voltammetry, LSV)曲线Figure 3. Linear sweep voltammetry (LSV) curves for the HER (red) and the OER (blue) on practical (dashed) and ideal (solid) electrocatalysts, respectively
图 3 理想条件下和实际应用中,HER(红色实线)和OER(蓝色实线)的线性扫描伏安(Linear sweep voltammetry, LSV)曲线Figure 3. Linear sweep voltammetry (LSV) curves for the HER (red) and the OER (blue) on practical (dashed) and ideal (solid) electrocatalysts, respectively2.2 基于阳极替代反应的辅助电催化分解水制氢策略
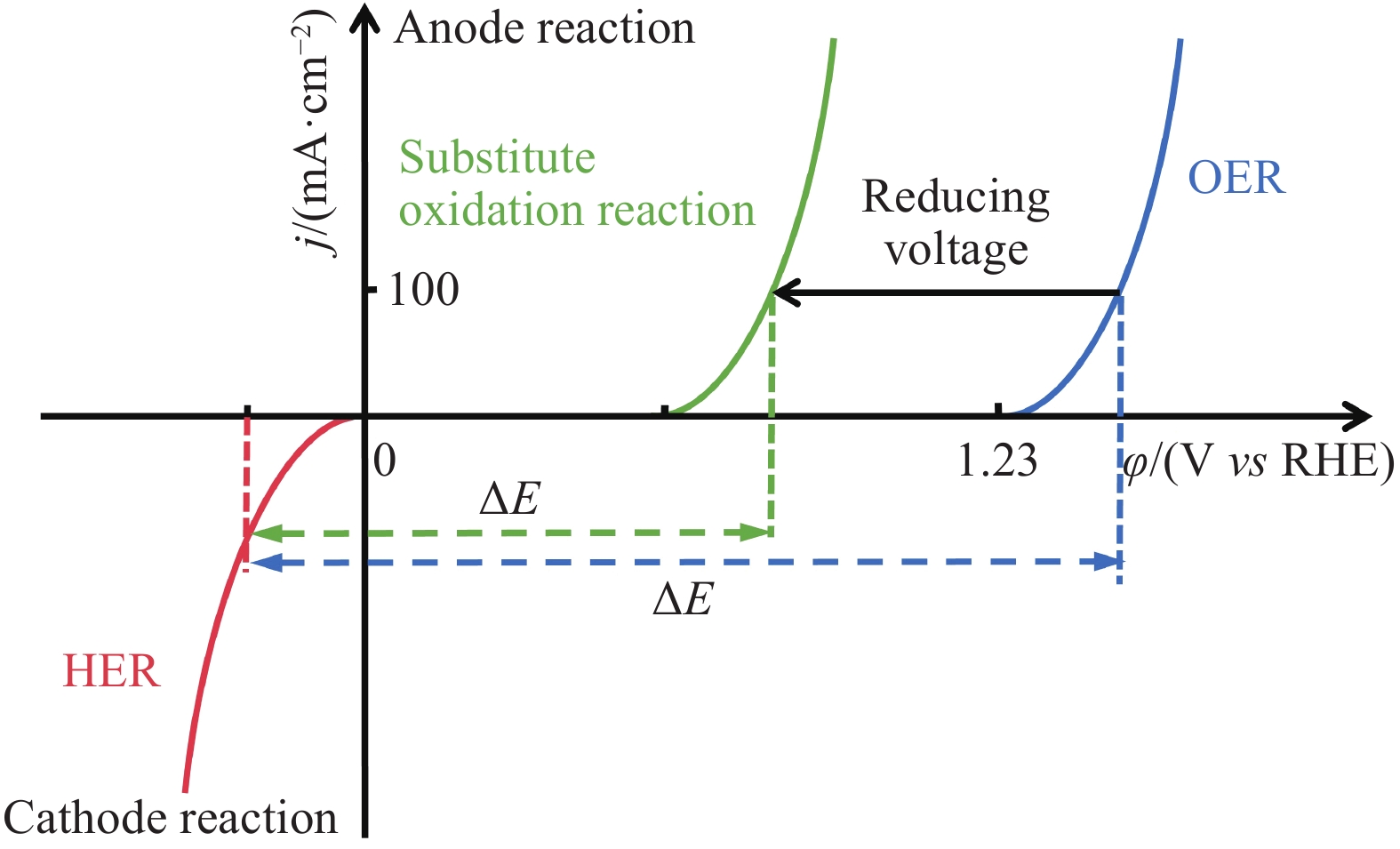
图4给出了基于阳极替代反应的电催化分解水制氢策略的原理. 其中,绿线表示阳极替代反应的极化曲线,其热力学平衡电位明显低于OER热力学平衡电位. 合适的阳极替代反应应遵循热力学平衡电位低于OER,不与OER发生竞争,其反应物在室温下易溶于水,其氧化产物不会影响HER的原则[16]. 此外,阳极替代反应的反应物应来源广泛,能够被电氧化降解或具有转换为高附加值化学品的潜力,进而提高电解水的经济效益. 因此,基于醇类、醛类、尿素、水合肼和氨的氧化反应替代OER辅助电解水制氢成为未来研究的新方向[17−18].
![]() 图 4 热力学平衡电位低于OER的阳极替代反应、HER和OER的LSV曲线Figure 4. LSV curves for anodic substitute reactions, HER, and OER
图 4 热力学平衡电位低于OER的阳极替代反应、HER和OER的LSV曲线Figure 4. LSV curves for anodic substitute reactions, HER, and OER3. 非贵过渡金属基高效催化剂的研究进展
3.1 非贵过渡金属基HER催化剂的研究进展
HER的本质是H在催化剂表面连续的吸脱附过程. 依据Sabatier原则[19],理想的HER催化剂既要与*H形成足够强的键,促进电子转移过程;又要确保在脱附过程中化学键容易断裂释放H2. 这意味着H在催化剂表面的吸脱附近似可逆. 因此,氢吸附自由能变化(ΔG*H)成为评价催化剂性能的重要指标[20]. Nørskov等[20]通过密度泛函理论(Density functional theory,DFT)计算发现ΔG*H为0的催化剂具有最佳的催化活性. 大量研究发现,非贵过渡金属基催化剂在HER中展现出了巨大的潜力,在某些条件下能够与ΔG*H接近于0的Pt等贵金属相媲美[21],但总体上其催化活性与贵金属相比仍存在差距,主要有以下策略降低非贵过渡金属基催化剂的HER过电位.
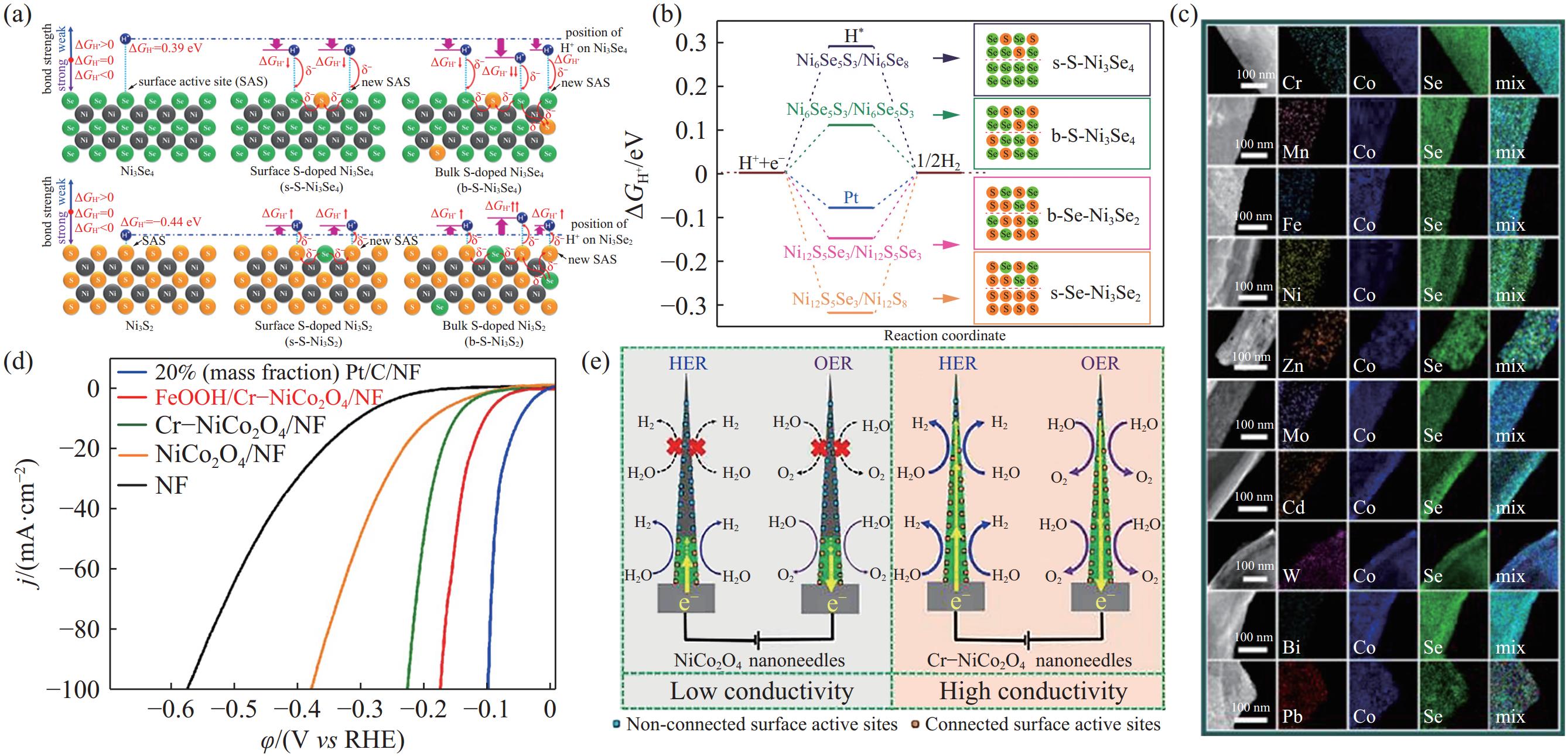
(1)掺杂元素:其他元素的掺入能够调控本征催化剂材料的电子结构[21],改变其活性位点附近的配位环境,最终改善催化剂对*H的吸脱附行为. 对于纯相的Ni3Se4和Ni3S2,其本征催化活性差,因此Liu等[22]制备了体相S掺杂的Ni3Se4和体相Se掺杂的Ni3S2催化剂,提高了催化位点的本征活性,降低了Ni3Se4和Ni3S2的ΔG*H,如图5(a)~(b) 所示. 对于CoSe2催化剂,S元素的掺杂能够优化催化剂活性位点的电子结构和配位环境,促使ΔG*H更接近于0,加快析氢反应速度[23]. 在DFT计算中通常以电子伏特(eV)为ΔG*H的单位,其中1 eV=
96485 J·mol−1. Zhang等[24]研发的S掺杂M-CoSe1.28S0.72催化剂达到10 mA·cm−2的电流密度仅需67 mV的过电位,并能够在酸性电解质溶液中稳定运行1000 h. 除了非金属元素以外,金属元素也能够作为掺杂剂提升催化剂的本征催化活性. Wu等[25]将Pb等10种单原子分别掺入CoSe2复合的二乙烯三胺(CoSe2–DETA)纳米带(图5(c)),掺入的单原子影响了Co的配位环境,提升了CoSe2的催化活性. 此外,元素掺杂可以提高催化剂的导电性,使其表面更多的活性位点与基底电极导通,增加有效催化位点的数量[26]. 例如高长径比的NiCo2O4纳米针,其导电性较差,整体催化活性不高. 将Cr元素掺入NiCo2O4纳米针中,纳米针的导电性大幅提高,与电极导通的活性位点的数量大大增加(图5(d)~(e)),HER性能显著提升[26].![]() 图 5 阴离子体相掺杂和表面掺杂的S‒Ni3Se4和Se‒Ni3S2的(a) *H吸附示意图和(b) 活性位点的ΔG*H[22]; (c) 单原子掺杂CoSe2–DETA纳米带的能谱[25]; (d) NiCo2O4和Cr‒NiCo2O4 纳米针的析氢LSV曲线及(e) 纳米针中电化学活性区域和惰性区域示意图[26].Figure 5. (a) Hydrogen adsorption scheme and (b) ΔG*H for bulk- and surface-doped S‒Ni3Se4 and Se‒Ni3S2[22]; (c) elemental mapping images of CoSe2–DETA nanobelts doped with different metallic single atoms[25]; (d) LSV curves for the HER on pure NiCo2O4 and Cr-doped NiCo2O4; (e) scheme of poorly conductive sites on NiCo2O4 nanoneedles and their activation for the HER from Cr-regulated conductivity[26]
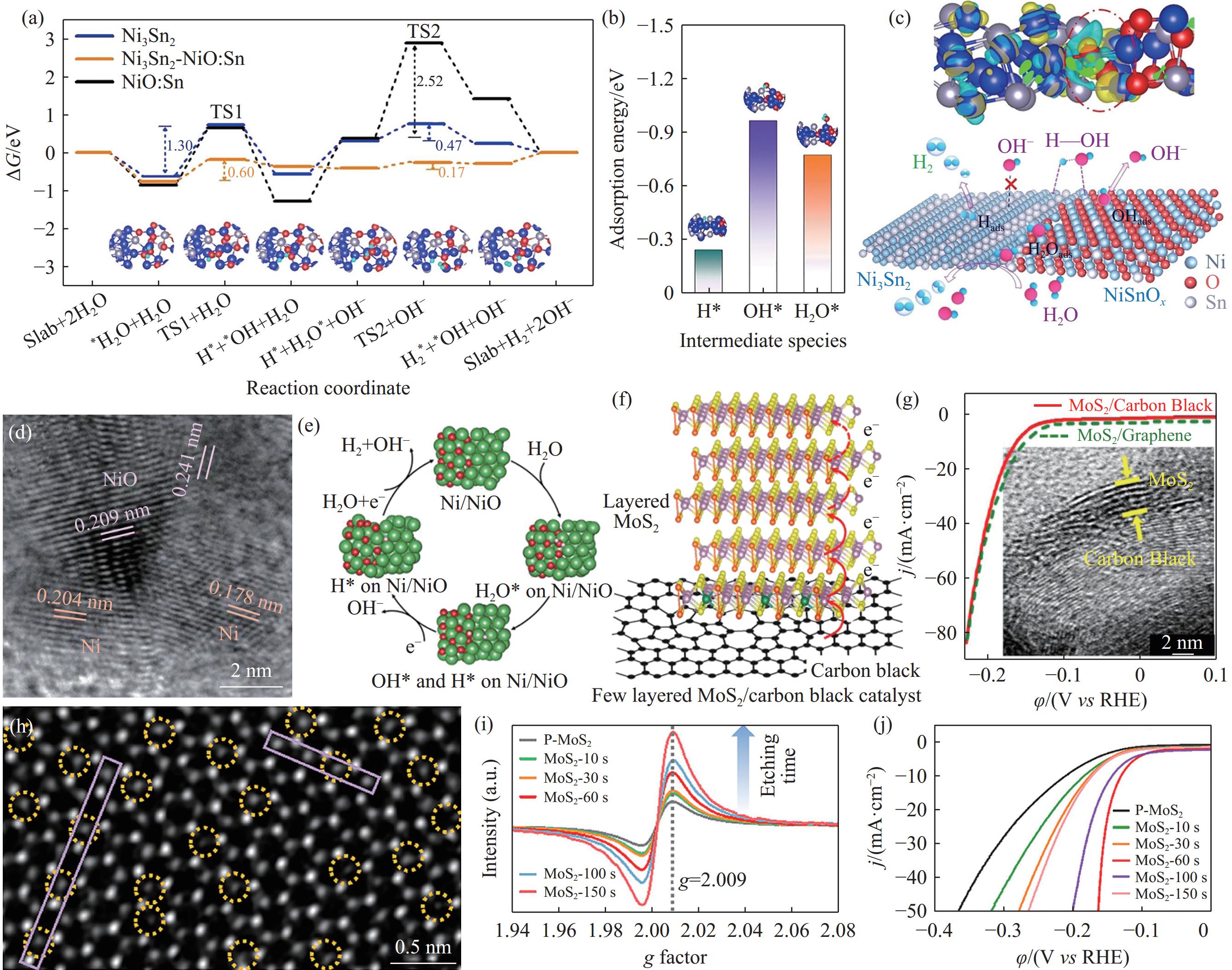
图 5 阴离子体相掺杂和表面掺杂的S‒Ni3Se4和Se‒Ni3S2的(a) *H吸附示意图和(b) 活性位点的ΔG*H[22]; (c) 单原子掺杂CoSe2–DETA纳米带的能谱[25]; (d) NiCo2O4和Cr‒NiCo2O4 纳米针的析氢LSV曲线及(e) 纳米针中电化学活性区域和惰性区域示意图[26].Figure 5. (a) Hydrogen adsorption scheme and (b) ΔG*H for bulk- and surface-doped S‒Ni3Se4 and Se‒Ni3S2[22]; (c) elemental mapping images of CoSe2–DETA nanobelts doped with different metallic single atoms[25]; (d) LSV curves for the HER on pure NiCo2O4 and Cr-doped NiCo2O4; (e) scheme of poorly conductive sites on NiCo2O4 nanoneedles and their activation for the HER from Cr-regulated conductivity[26](2)构建异质结构:为了改善NiSx对*H的吸脱附行为,Lin等[27]构建了NiSx–WO2.9异质结构,其相界面处的强电子相互作用加速了电子转移过程提高了HER活性. 为了增加过渡金属基硫属化合物活性位点的数量及本征催化活性,Zhang等[28]构筑了W掺杂的立方相/正交相CoSe2催化剂,相界面处形成了高活性的Co—Se—W活性物种,能够与Pt的性能相媲美. 为了加速碱性HER中H2O的解离过程,Wang等[29]制备了Ni3Sn2–NiSnOx异相结构的碱性HER催化剂. Ni3Sn2相具有理想的ΔG*H(图6(a)~(b)),而NiSnOx相能够促进H2O的解离(图6(c)). Li等[30]在多孔石墨碳基底(Porous graphitic carbon, PGC)上构建了Ni–NiO异质结构(图6(d)). 相比于Ni和NiO,Ni–NiO的异质结构不仅促进了H2O的解离(图6(e)),同时加快了电荷转移速率并增加了有效活性位点的数量.
![]() 图 6 (a) Ni3Sn2、Sn掺杂NiO(NiO:Sn)和Ni3Sn2–NiO:Sn吉布斯自由能能垒图,其中TS1表示过渡态1,TS2表示过渡态2;Ni3Sn2-NiSnOx (b)对中间物种的吸附能; (c) 电荷密度分布以及HER机理示意图[29]. (a)~(c) 中蓝色、红色、灰色和天蓝色的小球分别代表Ni, O, Sn和H原子. Ni–NiO/PGC的 (d) 高分辨透射(HRTEM)图; (e) Ni活性位点的HER机理示意图[30]. MoS2/CB的 (f) 结构示意图; (g) LSV曲线和HRTEM图[31]. 在H2O2中刻蚀不同时间的MoS2-x s(x=10~150)(h) MoS2-60 s的STEM图; (i) 电子顺磁共振(EPR)谱; (j) HER的LSV曲线[34]Figure 6. (a) Energy profiles for the HER on Ni3Sn2, Sn-doped NiO (NiO: Sn) and Ni3Sn2–NiO: Sn; TS1: transition state 1, TS2: transition state 2; (b) adsorption energies of intermediate species and (c) charge density difference for the HER on Ni3Sn2–NiSnOx[29]. Blue, red, gray, and small sky-blue spheres represent Ni, O, Sn, and H atoms, respectively. (d) HRTEM images and (e) HER mechanism on the Ni–NiO heterostructure[30]. (f) Structure of and (g) HER LSV curves for the MoS2/CB[31]. (h) STEM image for MoS2-60 s. (i) EPR spectra and HER LSV curves for MoS2-x etched for different times in H2O2 (x=10–150)[34]
图 6 (a) Ni3Sn2、Sn掺杂NiO(NiO:Sn)和Ni3Sn2–NiO:Sn吉布斯自由能能垒图,其中TS1表示过渡态1,TS2表示过渡态2;Ni3Sn2-NiSnOx (b)对中间物种的吸附能; (c) 电荷密度分布以及HER机理示意图[29]. (a)~(c) 中蓝色、红色、灰色和天蓝色的小球分别代表Ni, O, Sn和H原子. Ni–NiO/PGC的 (d) 高分辨透射(HRTEM)图; (e) Ni活性位点的HER机理示意图[30]. MoS2/CB的 (f) 结构示意图; (g) LSV曲线和HRTEM图[31]. 在H2O2中刻蚀不同时间的MoS2-x s(x=10~150)(h) MoS2-60 s的STEM图; (i) 电子顺磁共振(EPR)谱; (j) HER的LSV曲线[34]Figure 6. (a) Energy profiles for the HER on Ni3Sn2, Sn-doped NiO (NiO: Sn) and Ni3Sn2–NiO: Sn; TS1: transition state 1, TS2: transition state 2; (b) adsorption energies of intermediate species and (c) charge density difference for the HER on Ni3Sn2–NiSnOx[29]. Blue, red, gray, and small sky-blue spheres represent Ni, O, Sn, and H atoms, respectively. (d) HRTEM images and (e) HER mechanism on the Ni–NiO heterostructure[30]. (f) Structure of and (g) HER LSV curves for the MoS2/CB[31]. (h) STEM image for MoS2-60 s. (i) EPR spectra and HER LSV curves for MoS2-x etched for different times in H2O2 (x=10–150)[34](3)复合碳基材料:典型的层状MoS2和MoSe2催化剂层间电阻远高于层内电阻,阻碍了HER中电子传递的过程. Xue等[31−32]在炭黑(Carbon black, CB)表面上生长层数少、尺寸小的MoS2和MoSe2(图6(f)~(g)). 实验表明MoS2/CB和MoSe2/CB在HER过程中的电荷转移电阻大幅降低,同时暴露了更多的边缘活性位点,有效地降低了HER过电位. 相比于炭黑、碳纳米管等,碳点尺寸小易于修饰功能化官能团,为电解质溶液与催化剂的活性位点之间提供理想的界面. 因此,Song等[33]采用含氮量不同的前驱体合成了三种碳点并负载在磷化钼(MoP)纳米颗粒上. 经氮掺杂的碳点有效提高了MoP的导电性,氮的引入优化了活性位点对*H的吸脱附行为,提升了MoP的催化活性.
(4)缺陷工程:向材料晶格中引入缺陷,将对材料晶格表面的组分、电子结构、电荷分布及局部环境产生影响. MoS2的活性位点通常位于其片层边缘,为增加催化活性位点数量,Wang等[34]通过化学刻蚀法成功向MoS2片层内中引入了孤立的S空位(图6(h)~(i)),并在电子顺磁共振波谱中检测到S空位的信号,对应的波谱分裂因子g为2.009,该峰值强度与MoS2中S空位浓度成正比. 在含有S空位的MoS2中,邻近空位的电子结构发生改变,在电催化惰性的MoS2片层内引入新的活性位点,将原来产生10 mA·cm−2电流密度需要220 mV的过电位降低至110 mV(图6(j)). Jin等[35]通过引入S空位,优化了NiS2的电子结构与配位环境,当空位浓度为5.9%时,该催化剂的ΔG*H接近于0,10 mA·cm−2电流密度下的析氢过电位相比于NiS2降低了74 mV. Zhan等[36]从理论计算和实验两方面证明了在S空位周围暴露更多的Mo原子有助于MoS2的本征催化活性提升,阐释了空位周围原子环境与催化活性之间的联系,为制备高效的HER催化剂提供了新的参考.
非贵过渡金属基HER催化剂种类繁多,主要包括过渡金属基硫属化合物、磷化物,过渡金属基氢氧化物和氧化物等催化剂. 每类催化剂活性中心的化学环境和电子结构不尽相同,因此针对不同类别的HER电催化剂,提高催化剂活性的策略也有所差别. 前述提高HER催化剂活性的不同策略主要针对不同类型的催化剂,指向性地改善催化活性中心对HER中间产物的吸脱附行为,增加有效催化活性位点的数量. 对于过渡金属基氢氧化物、氧化物类HER催化剂,其活性位点催化活性较低[22−25,27−28],掺杂和构建异质结构可以有效改善此类催化剂活性位点的电子结构和配位环境[21,37]. 对于过渡金属基硫属化合物、磷化物类型的催化剂导电性差[31−32],其有效催化活性位点数量少,而复合高导电率的碳基材料能够提高该类催化剂的导电性增强并暴露出更多的活性位点[32];对于层状过渡金属基硫化物类型的催化剂而言,该类催化剂片层边缘具有催化活性,片层内部则为催化惰性,通常在催化剂片层内部构建缺陷增加新的活性位点,促使催化剂从面内惰性转变为面内活性[34].
3.2 非贵过渡金属基OER催化剂的研究进展
相比于HER过程,OER动力学过程迟缓,成为电解水反应中电能消耗的主要来源. 在AEM和LOM机制中,OER的反应过电位由各步骤中反应自由能(ΔG)最大的限速步骤(Rate determined step,RDS)决定[38]. Man等[38]发现ΔG*OH和ΔG*OOH存在比例关系,并建立了(ΔG*O–ΔG*OH)与OER性能之间的“火山型”曲线. 除了热力学理论计算外,Lin等[39−40]通过实验测得了Arrenius形式的活化能,为评估OER催化剂的动力学能垒提供了新的思路. 尽管RuO2和IrO2作为商用催化剂展现出了优异的OER性能,但是Ir、Ru贵金属储量匮乏,在工况下存在贵金属溶出现象,导致其催化活性下降. 非贵过渡金属基材料具有价格低廉、组分易调控、结构稳定性高、电阻率小等优势而被视为IrO2、RuO2催化剂的有力竞争者. 为了不断优化和提升非贵过渡金属基催化剂的OER性能,现有研究中有效降低OER过电位的催化剂设计策略如下.
(1)元素掺杂:单金属氢氧化物催化剂对于OER中间体的吸脱附性能不佳,因此Lu等[41]合成了镍铁层状双金属氢氧化物(NiFe layered double hydroxide, NiFe LDH). 伴随着Fe元素的掺入,催化剂的OER活性大幅提高,达到30 mA·cm−2电流密度所需过电位从原有450 mV降低至280 mV. 研究人员发现向过渡金属氧化物中掺入其他元素,可改善活性中心的吸脱附行为和成键强度. Li等[42]制备了Cr掺杂非晶相的CoCrOx,其中Cr3+的掺入缩短了Co—O键的键长,优化了Co活性位点的配位环境,DFT计算表明其RDS步骤能垒显著降低. Chong等[43]合成了La和Mn共掺杂的钴尖晶石型酸性OER催化剂. La3+和Mn3+分别增强了Co3O4的耐酸性和导电性. 该催化剂在2.47 V的槽压下能够达到2000 mA·cm−2的电流密度,其性能已接近于商用的Ir催化剂.
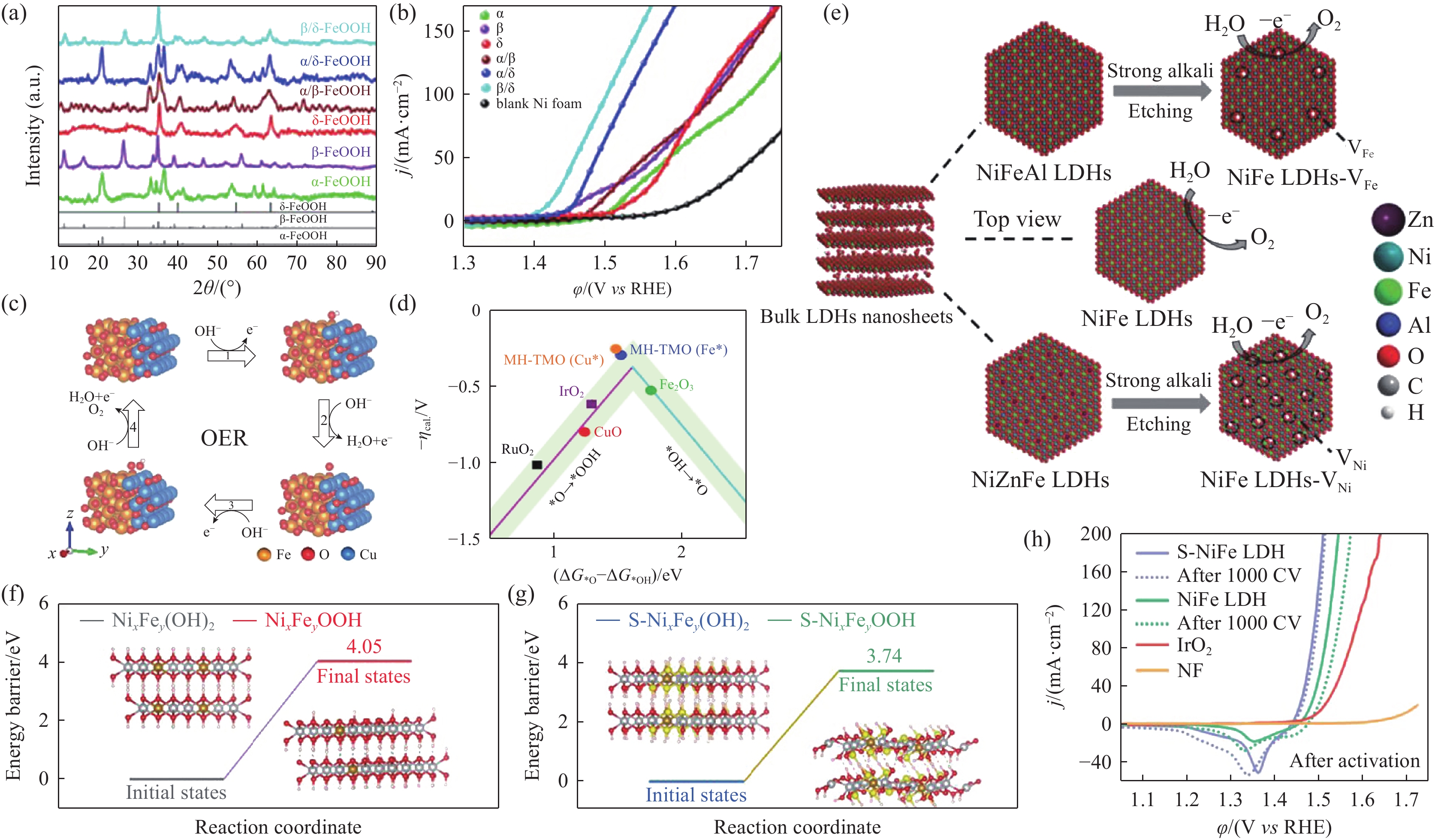
(2)调控晶体结构:晶体结构的改变通常会影响材料的电子结构,从而引起其电催化活性的改变. Lin等[39]分别在镍网(nickel foam, NF)上制备了β相NiFeOOH/NF和γ相NiFeOOH/NF. 相比于宽间距的γ相NiFeOOH/NF,紧密堆垛的β相NiFeOOH/NF具有更小的OER活化能能垒. Hu等[44]利用含有不同阴离子的前驱体溶液,通过溶剂热反应分别制备了单相(α、β和δ)和混合相(α/β、α/δ和β/δ)的FeOOH催化剂(图7(a)). DFT计算结果表明β/δ混合相的FeOOH中存在更多的氧空位,这些氧空位优化了FeOOH的电子结构,从而降低了*OH氧化为*O中间步骤的反应能垒,展现出最为优异的OER性能(图7(b)).
![]() 图 7 α、β、δ、α/β、α/δ和β/δ相的FeOOH (a) XRD图; (b) OER的LSV曲线[44]; (c) Fe2O3–CuO(Fe 位点)的OER过程示意图; (d) 过电位负值与ΔG*O–ΔG*OH之间的火山型关系图[47]; (e) NiFe LDH-VNi和NiFe LDH-VFe催化剂示意图[50]; (f) NixFey(OH)2和 (g) S-NixFey(OH)2表面重构脱氢能垒图; (h) 不同催化剂OER的LSV曲线[57]Figure 7. (a) XRD patterns and (b) LSV curves for the OER on α, β, δ, α/β, α/δ, and β/δ phases FeOOH[44]; (c) scheme of the OER on the Fe2O3–CuO (Fe site) heterostructure; (d) Overpotentials plotted against a descriptor of ΔG*O–ΔG*OH[47]; (e) Scheme of NiFe LDH-VNi and NiFe LDH-VFe synthesized by alkali etching of the LDHs[50]; Energetic profiles for dehydrogenation on (f) NixFey(OH)2 and (g) S-NixFey(OH)2; (h) LSV curves of OER for different catalysts[57].
图 7 α、β、δ、α/β、α/δ和β/δ相的FeOOH (a) XRD图; (b) OER的LSV曲线[44]; (c) Fe2O3–CuO(Fe 位点)的OER过程示意图; (d) 过电位负值与ΔG*O–ΔG*OH之间的火山型关系图[47]; (e) NiFe LDH-VNi和NiFe LDH-VFe催化剂示意图[50]; (f) NixFey(OH)2和 (g) S-NixFey(OH)2表面重构脱氢能垒图; (h) 不同催化剂OER的LSV曲线[57]Figure 7. (a) XRD patterns and (b) LSV curves for the OER on α, β, δ, α/β, α/δ, and β/δ phases FeOOH[44]; (c) scheme of the OER on the Fe2O3–CuO (Fe site) heterostructure; (d) Overpotentials plotted against a descriptor of ΔG*O–ΔG*OH[47]; (e) Scheme of NiFe LDH-VNi and NiFe LDH-VFe synthesized by alkali etching of the LDHs[50]; Energetic profiles for dehydrogenation on (f) NixFey(OH)2 and (g) S-NixFey(OH)2; (h) LSV curves of OER for different catalysts[57].(3)构建异质结构:异质结构的相界面是由两种或多种组分构成的,界面耦合能够优化不同组分的电子结构. 为了提高氧化物的导电性,He等[45]构建了Ni2Co1@Ni2Co1Ox的核壳结构,以Ni2Co1金属作为核心大幅提高了Ni2Co1Ox的导电性,从而提高了OER活性. 而单一组分对OER中间产物的吸脱附能力有限,因此Zhao等[46]制备了CoNi LDH–FeOOH异相催化剂,实验和理论计算结果表明OER中间产物在相界面处的吸脱附行为得到改善,其OER限速步骤的能垒降低. Hu等[47]合成的Fe2O3–CuO催化剂,在相界面处形成了Fe—O—Cu键,优化了OER中间产物在Fe和Cu位点的吸脱附行为,图7(c)中的1~4中间产物吸脱附步骤对应于1.2节中的式(9)~(11). 在DFT计算得到的“火山型”曲线中(图7(d)),Fe2O3–CuO位于Fe2O3和CuO的中间,具有最理想的催化性能. Huang等[48]制备了Co3O4–CeO2催化剂,在引入CeO2相后,Co3O4的界面成键环境发生改变,Co3+更容易被氧化为高催化活性的Co4+,从而降低了反应过电位.
(4)缺陷工程:缺陷工程是通过精确控制缺陷的种类和浓度,针对性地破坏催化剂材料的晶格周期性从而提升本征材料的催化性能[49]. 为了进一步开发NiFe LDH的催化潜力,Wang等[50]利用强碱刻蚀法制备了含有Ni2+空位的NiFe LDHs-VNi和含有Fe3+空位的NiFe LDHs-VFe,如图7(e) 所示. 实验和理论计算结果表明丰富的Ni和Fe空位有效地调节了催化剂表面的电子结构,优化了中间产物的吸脱附行为,降低了OER过电位. Xu等[51]通过制造O空位缺陷,提高了Co3O4的导电性,增加了电化学活性面积,降低了催化剂的OER过电位. 此外,缺陷的引入在提升催化活性的同时,还在一定程度上提高了催化剂的稳定性. Peng等[52]研究发现阳离子空位增加了邻近金属与O的结合能,减少了晶格畸变,阻止了NiFe LDH在高过电位氧化过程中金属元素的溶出. 因此,富含Ni2+和Fe3+空位的NiFe LDH催化剂同时实现了高催化活性和长期稳定性.
(5)复合碳基材料:非贵过渡金属基硫化物在OER中存在耐久性差的问题,Wu等[53]合成氮掺杂碳点修饰的FeCoSy的纳米片,不仅提高了FeCoSy的长期循环稳定性和导电性,同时增大了催化剂的电化学活性面积,暴露出更多的活性位点. 类似地,Fe、Co、Ni等非贵金属在强碱性和高过电位条件下也面临着循环稳定性差和OER活性低的问题,因此Cui等[54]将纳米Fe、Co和Ni颗粒封装在单层石墨烯中. 实验结果表明由单层石墨烯构建的M@NC结构激活了石墨烯表面的惰性位点. 为进一步提高原子利用率和本征催化活性,研究人员利用碳载体构建了Co—N—C单原子活性位点[55]. 此外,Li等[56]通过ZIF-67热解合成了氮碳层包埋Co纳米颗粒的催化剂,暴露出更多的活性位点,实现了催化活性的提升.
(6)表面重构:大多数过渡金属基催化剂(过渡金属氢氧化物、过渡金属硫属化合物、过渡金属磷化物、过渡金属氮化物等)在电化学氧化条件下都会经历表面溶解再沉积的重构过程[58]. 例如,Lei等[57]制备的S-NiFe LDH在OER过程中发生表面重构. 实验和理论计算表明S掺杂有效地降低了NixFey(OH)2氧化为NixFeyOOH的重构能能垒(图7(f)~(g)),生成的S-NixFeyOOH作为活性相改善了OER中间产物的吸脱附行为,降低了OER限速步骤的能垒,表现出优异的催化活性(图7 (h))和长期稳定性. 类似地,Huang等[59]研究发现P掺杂的FeSe2纳米棒在高过电位氧化过程中表面重构为富含Se空位的P-FeOOH,而FeOOH作为催化活性相显著提高了FeS2的催化活性.
非贵过渡金属基OER催化剂包括非贵过渡金属合金,过渡金属基氢氧化物、氧化物、磷化物、硫化物等,每类OER催化剂具有独特的组分和结构特性,需要对特定的催化活性中心进行优化,针对性地提升催化剂的催化活性. 对于过渡金属基氧化物、氢氧化物类OER催化剂,其本征催化活性低[60],掺杂元素、调控晶体结构和构建异质结构能够改善OER中间产物的吸脱附行为,是提高该类催化剂活性的核心策略[39,41,49];对于合金、过渡金属基氢氧化物、硫属化合物类催化剂,其导电性差,复合炭黑、碳点等碳材料能够增强其导电性并暴露出更多的活性位点[61];对于过渡金属基硫磷化合物类催化剂,在电化学氧化条件下诱导其表面重构是提高该类催化剂活性的核心策略[58].
4. 基于阳极替代反应的辅助电催化分解水制氢策略
已开发的HER和OER催化剂能够大幅降低过电位,但受限于OER的高能量需求,电解水过程仍然需要大于1.8 V的槽压才能产生可观的电流密度[62]. 因此,选择合适的阳极氧化反应替代OER是提高电解水制氢效率的有效策略[63]. 现有研究表明,以尿素、水合肼、氨、醇、醛和硫等作为反应物的氧化反应能够有效替代OER,降低电解水制氢能耗[16,64]. 尽管阳极替代反应具有较低的热力学平衡电位,但是反应物的氧化过程通常涉及多电子转移,导致反应动力学缓慢. 因此,基于上述阳极替代反应设计高催化活性的催化剂是降低电解水制氢电位的核心. 本文以醇类和含氮化合物两类常见反应物的氧化反应为例,总结非贵过渡金属基催化剂在降低阳极替代反应过电位中的应用.
4.1 醇类氧化反应
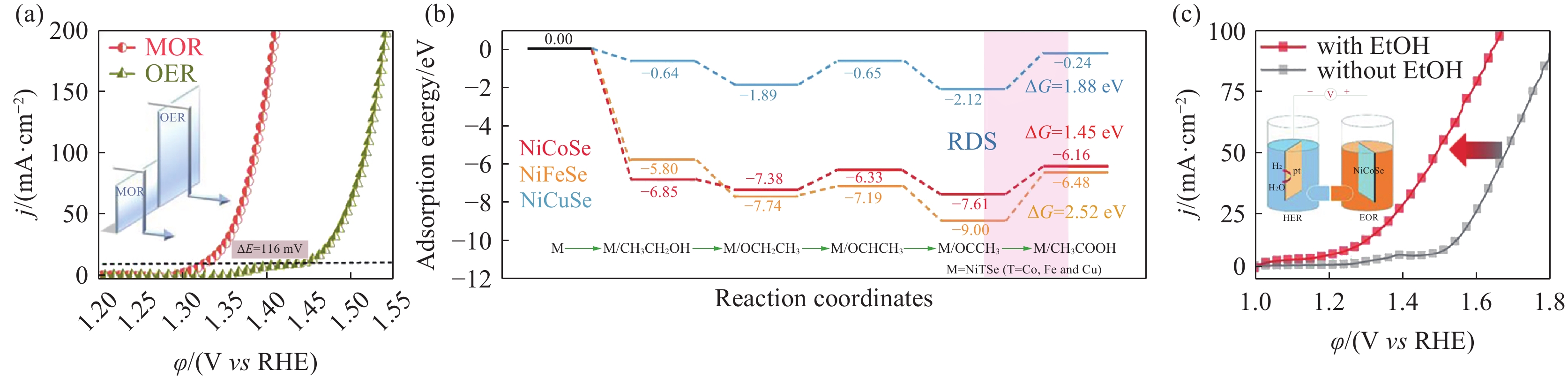
醇类氧化反应(Alcohol oxidation reactions, AORs)的热力学平衡电位低于OER,醇类反应物在电化学氧化过程中易被氧化为醛或酸. Zhao[65]等在碳纸(carbon cloth, CC)上合成了以NiSe为核,碳纳米纤维(carbon nanofibers, CNFs)为壳的CNFs@NiSe/CC催化剂,在甲醇氧化反应(Methanol oxidation reaction, MOR)辅助电解水制氢中,CNFs@NiSe/CC作为双功能催化剂,阴极析氢的速率为35.67×10−8 mol·s−1,是替代OER前产氢速率的7.5倍. Hao等[66]通过构建Fe2O3–NiO异质催化剂,降低了MOR限速步骤的能垒(图8(a)). 在MOR辅助制氢中,该体系产生10 mA·cm−2的电流密度仅需1.38 V,远低于电解水中1.52 V的电压. 此外,阳极产物中没有O2的存在有力地证明了MOR完全替代了OER. Li[67]等制备了NiCuSe、NiFeSe和NiCoSe催化剂,通过实验和DFT计算表明NiCoSe具有最优的电子结构,降低了乙醇氧化反应(Ethanol oxidation reaction, EOR)限速步骤的能垒(图8 (b)). 在EOR辅助制氢中,以在碳纸负载的Pt/CC及NiCoSe催化剂作为阴、阳极催化剂,产生50 mA·cm−2电流密度所需的电压比电解水制氢的电压降低了180 mV(图8(c)). Xia等[68]制备的NiCrO-VCr,O催化剂在甘油氧化中的起峰过电位仅为1.32 V,远低于OER中的1.55 V. 以上研究结果表明AORs能够有效替代OER降低整体电解水的反应电位.
4.2 含氮类化合物氧化反应
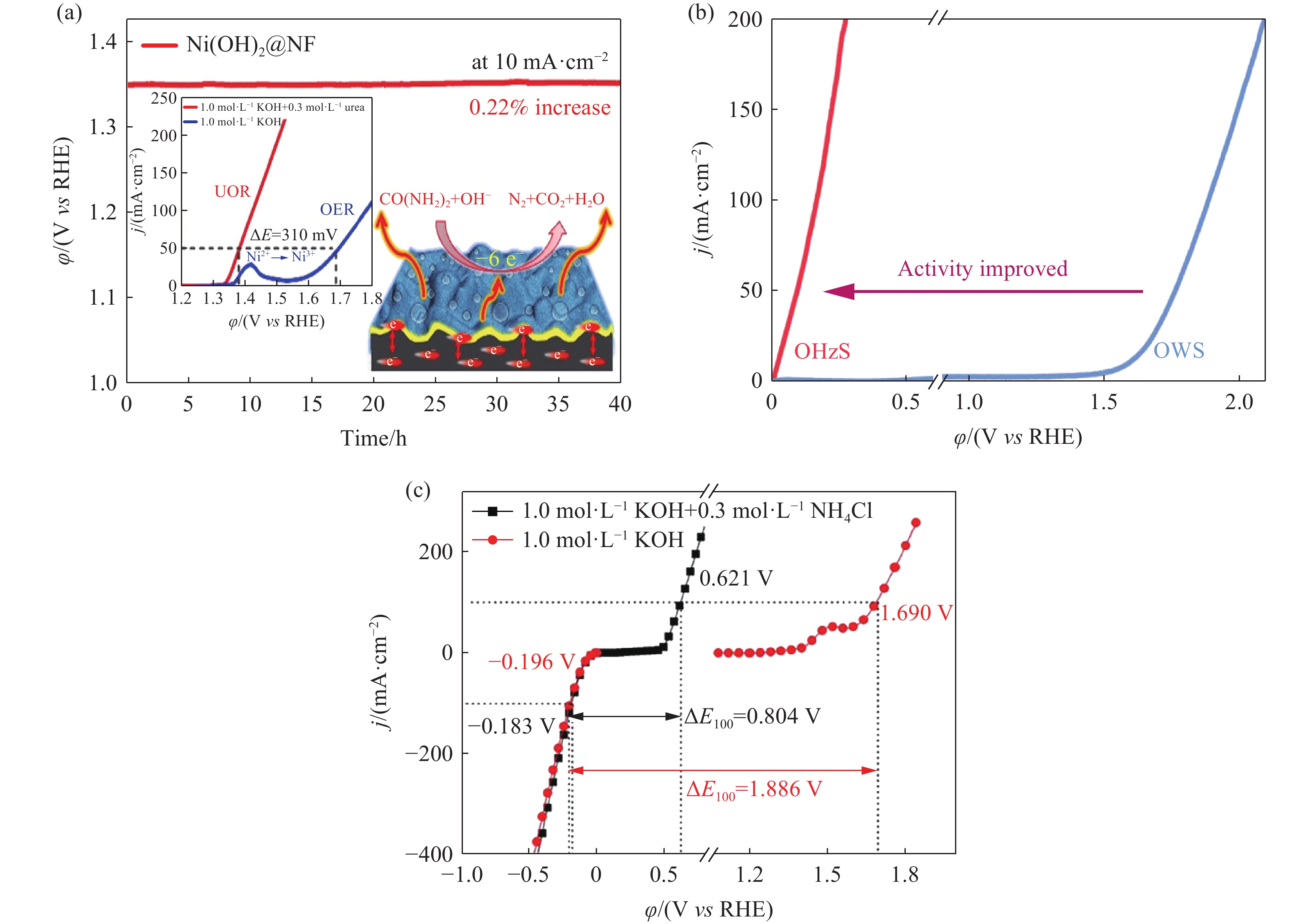
含氮类化合物主要包括尿素(CO(NH2)2)、水合肼(N2H4)和氯化铵(NH4Cl). 尿素氧化反应(Urea oxidation reaction, UOR)的热力学平衡电位为0.37 V vs RHE,远低于OER的热力学平衡电位[69]. 然而,UOR过程涉及六电子转移,需要利用高效的催化剂提高反应速率. Xia等[70]制备了超薄多孔的Ni(OH)2/NF,在UOR辅助制氢中,Pt/C和Ni(OH)2/NF分别作为阴、阳极催化剂,仅需1.45 V的电压就能提供50 mA·cm−2的电流密度,并稳定运行超过40 h. 然而在电解纯水制氢中,需要额外增加0.31 V的电压才能够提供相同的电流密度(图9(a)). 为了进一步提高催化剂的UOR活性,Du等[71]构建了CoS1.097–Ni3S2的异质结构. 在相界面处,两相分别吸附尿素分子中的氨基和羰基,加速了尿素的分解. 水合肼氧化反应(Hydrazine oxidation reaction, HzOR)的热力学平衡电位为 −0.33 V vs RHE. Tang等[72]利用NiP/NF催化剂氧化水合肼辅助制氢,该体系中达到500 mA·cm−2的电流密度仅需1.0 V的电压,远低于电解纯水制氢所需的电压. Liu等[73]在NF利用P、W掺杂优化了Co3N纳米线阵列(PW-Co3N nanowire arrays, PW-Co3N NWA/NF)的电子结构,促进了HER中的氢吸附和HzOR中的脱氢过程,提高了两电极产氢效率(图9(b)). 在HzOR辅助制氢中,达到200 mA·cm−2的电流密度仅需0.277 V的电压,与电解纯水制氢相比,节能率达86.8%. 在碱性条件下氨氧化反应(Ammonia oxidation reaction, AmOR)的热力学平衡电位为 −0.77V vs RHE,比UOR和HzOR的理论氧化电位更低. Hu等[74]在还原氧化石墨烯(reduced graphene oxide, rGO)上负载了具有HER和AmOR活性的N掺杂的NiZnCu LDH(N-NiZnCu LDH/rGO)催化剂. 如图9(c) 所示,该催化剂的AmOR电位显著低于OER电位,同时NH4Cl的引入不会影响HER过程. 在AmOR辅助制氢中,以N-NiZnCu LDH/rGO作为催化剂,仅需0.769 V的槽压就能提供50 mA·cm−2的电流密度,大幅降低了电能消耗.
![]() 图 9 (a) Ni(OH)2@NF氧化尿素的示意图,在有无尿素电解液中的LSV曲线以及稳定性[70]; (b) PW-Co3N NWA/NF催化剂组成的两电极在有无水合肼的电解液中的LSV曲线[73],其中,OHzS表示混合水合肼的水分解,OWS表示全水解; (c) N-NiZnCu LDH/rGO在有无0.3 mol·L−1 NH4Cl的1.0 mol·L−1 KOH电解液中的LSV曲线[74]Figure 9. (a) Scheme for urea oxidation, together with the LSV curves with and without urea electrolyte, and the chronopotentiometry plot for Ni(OH)2@NF catalyst[70]; (b) LSV curves of PW-Co3N NWA/NF as both the anode and cathode with and without the addition of hydrazine[73]; Overall hydrazine splitting is denoted as OHzS and overall water splitting is denoted as OWS; (c) LSV curves of N-NiZnCu LDH/rGO in 1.0 mol·L−1 KOH with and without 0.3 mol·L−1 NH4Cl[74
图 9 (a) Ni(OH)2@NF氧化尿素的示意图,在有无尿素电解液中的LSV曲线以及稳定性[70]; (b) PW-Co3N NWA/NF催化剂组成的两电极在有无水合肼的电解液中的LSV曲线[73],其中,OHzS表示混合水合肼的水分解,OWS表示全水解; (c) N-NiZnCu LDH/rGO在有无0.3 mol·L−1 NH4Cl的1.0 mol·L−1 KOH电解液中的LSV曲线[74]Figure 9. (a) Scheme for urea oxidation, together with the LSV curves with and without urea electrolyte, and the chronopotentiometry plot for Ni(OH)2@NF catalyst[70]; (b) LSV curves of PW-Co3N NWA/NF as both the anode and cathode with and without the addition of hydrazine[73]; Overall hydrazine splitting is denoted as OHzS and overall water splitting is denoted as OWS; (c) LSV curves of N-NiZnCu LDH/rGO in 1.0 mol·L−1 KOH with and without 0.3 mol·L−1 NH4Cl[745. 电解水制氢的工程应用前景
尽管开发高效的催化剂及利用阳极替代反应辅助制氢的策略能够大幅降低电解水制氢的能耗,但是在实际的电解水工程应用中,电解槽装置、催化剂规模化的制备、工况条件以及商用的评价标准与基础研究之间存在差距,限制了电解水制氢技术在实际工程应用中的跨越式发展.
5.1 电解水制氢技术的发展现状
与实验室三电极体系不同,工业电解水制氢所采用的电解槽由阴/阳极催化剂、气体扩散层、隔膜和电解液组成. 目前,低温电解水制氢所使用的电解槽主要包括碱性电解槽(Alkaline water electrolyzer, AWE),质子交换膜电解槽(Proton exchange membrane water electrolyzer, PEMWE)和阴离子交换膜电解槽(Anion exchange membrane water electrolyzer, AEMWE),如图10(a)~(c) 所示[75]. 除高性能的催化剂以外,电解槽中膜、催化剂支撑材料、粘合剂、气体扩散层的性质和电解液局部 pH 值均会影响工业电解水制氢的效率以及氢气的纯度,如图10(d) 所示[75].
![]()
其中,AWE因其成本低廉,装置简单,是最常用的商用电解槽[76]. 商业应用中通常以Ni或者雷尼镍作为电极材料[77]. 最新研制的NiFe(OH)x–Ni3S2–Ni和Ni3Sn2–NiSnOx催化剂应用于两电极体系中,能够在1.53 V的槽压下,达到1 A·cm−2的商用电流密度,并稳定运行
3000 h[29]. 然而,AWE仍存在产氢效率低、电流密度低、欧姆电阻高、启停时间长等不足. 相比于AWE,PEMWE以质子交换膜为核心部件,高纯水为电解液,将阴/阳极电极材料组成了结构紧凑、电阻小的膜电极组件[78]. 其具有产氢效率高、电流密度高、启停响应快和氢气纯度高的优点. 为适应强酸性环境,商用PEMWE常以高成本的Pt作为HER催化剂,IrO2和RuO2作为OER催化剂. 目前,Chong等[43]将La、Mn掺杂的Co基催化剂和Nafion 115质子交换膜组装于PEMWE中,该器件在2.47 V的槽压下达到了2 A·cm−2的电流密度. 为克服质子交换膜成本高昂的问题,Wu等[79]以价格低廉的阴离子交换膜作为隔膜,以Cu0.7Co2.3O4作为阳极材料,该AEMWE在1.8 V的槽压下达到了1 A·cm−2的商业级电流密度. 总体上,AEMWE结合了AWE和PEMWE的优势,在降低电解槽成本的同时能够有效地提高电流密度和产氢效率,作为新兴的电解槽,现阶段仍处于实验室研究向工业应用转型阶段[80].5.2 电解水制氢的应用前景
目前,风电制氢和光伏发电制氢是最为典型的可再生能源制氢[81]. 单一的风电制氢或光伏发电制氢仍受限于风能和太阳能的间歇性和时空分布不均性,而开展风光互补电解水制氢,能够获得稳定的电能输出,提升电解水制氢的经济效益,具有广阔的应用前景. 丰富的海水资源能够为电解水制氢提供充足的原料,可降低对短缺的淡水资源的依赖,因此电解海水制氢作为新兴的制氢技术而备受关注[82−84]. 其中,直接电解海水制氢相比于海水纯化后制氢,能节省海水纯化设备及运输的成本,是极具发展潜力的制氢技术[85]. 直接海水电解技术的核心是设计开发耐腐蚀、高活性的催化剂及隔膜等组件以满足电解槽在海水中长期稳定的运行. 近期,Na等[86]制备了能够抵抗Cl−腐蚀的(Ni,Fe)O(OH)–NiCoS催化剂,在1 mol·L−1 KOH和天然海水的电解液中,组装的AEMWE能够在400 mA·cm−2和600 mA·cm−2的电流密度下稳定运行300 h,其电压衰减小于100 μV·h−1,为非贵过渡金属基催化剂用于直接海水电解技术提供了参考. 此外,向电解液中加入醇、醛、水合肼、尿素、胺等易氧化的电解质,替代水分子氧化,构建混合电解水体系,不仅能够减少电解水体系中电能的消耗,还能避免出现O2与H2交叉混合的安全隐患[87]. 然而,混合电解水的研究面临产物分离困难、是否适配现有的商用电解槽等诸多问题,尚处于起步阶段[88−89].
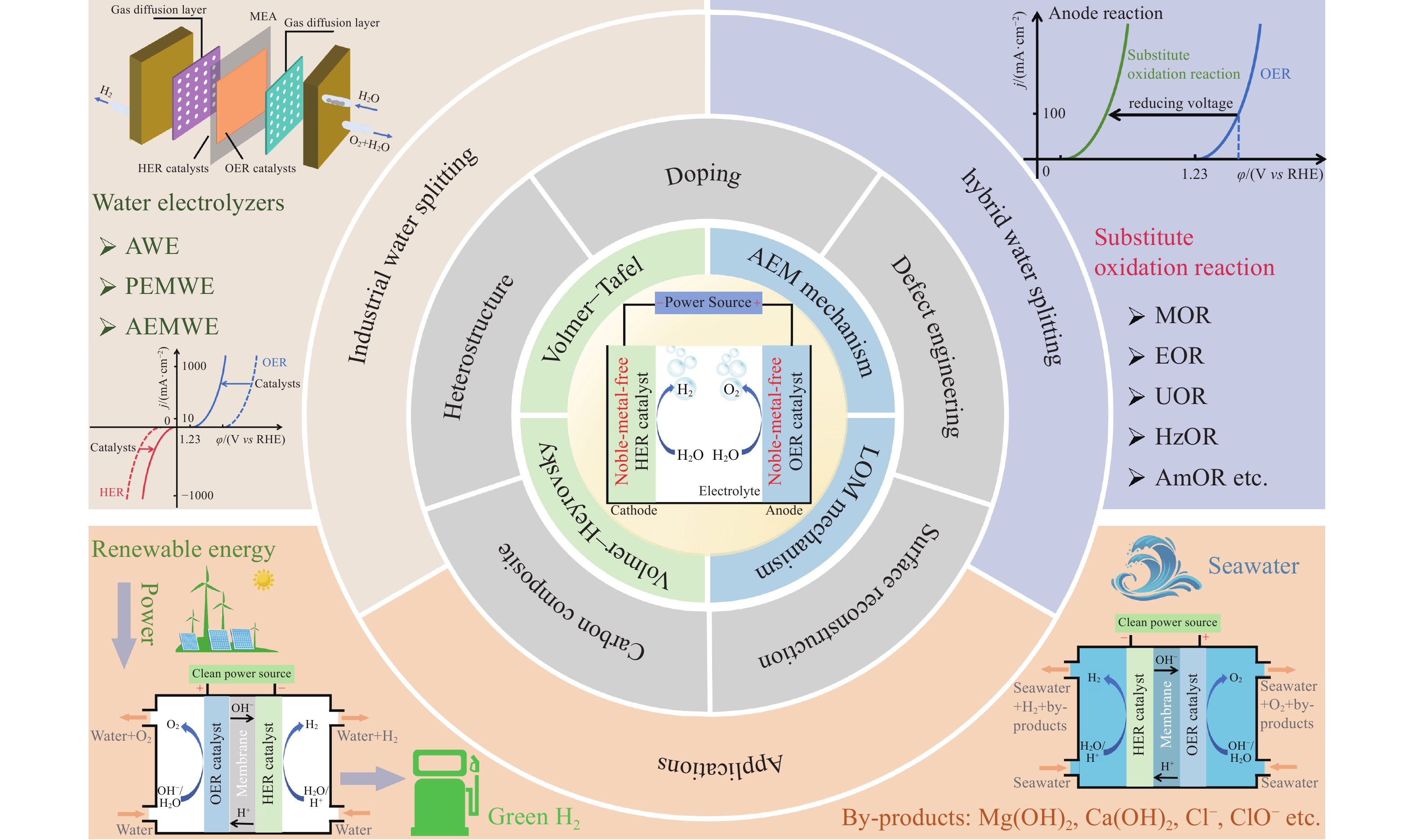
综上所述,本文从非贵过渡金属基催化剂参与HER和OER中的反应机理出发,归纳包含掺杂、异质结构、复合碳材料、缺陷工程和表面重构等提升催化剂性能的策略,并针对催化剂的研究现状提出阳极替代反应辅助制氢的策略,同时介绍电解水制氢的工程应用前景,如图11所示.
![]() 图 11 非贵过渡金属基催化剂参与HER和OER的反应机理、催化剂性能优化策略、阳极替代反应辅助制氢的策略以及电解水制氢的工程应用前景概述Figure 11. Overview of HER and OER mechanisms of noble-metal-free catalysts, optimization strategies, substitute oxidation reaction-assisted hydrogen production strategies, and engineering application prospects of hydrogen production by water splitting
图 11 非贵过渡金属基催化剂参与HER和OER的反应机理、催化剂性能优化策略、阳极替代反应辅助制氢的策略以及电解水制氢的工程应用前景概述Figure 11. Overview of HER and OER mechanisms of noble-metal-free catalysts, optimization strategies, substitute oxidation reaction-assisted hydrogen production strategies, and engineering application prospects of hydrogen production by water splitting6. 结论与展望
电解水在高效率、低能耗、零碳排放制氢上具有广阔前景. 开发低成本、高性能的非贵过渡金属基催化剂以及选择阳极氧化反应替代OER耦合制氢是提高制氢效率的重要手段. 本文总结了提升电极材料催化活性的主要策略以及可替代OER降低制氢电位的主要阳极氧化反应. 尽管上述策略能够在一定程度上降低制氢的能耗和成本,但在实际工业中实现大规模、清洁经济和可持续的电解水制氢,仍然面临许多问题和挑战.
(1)开发经济高效的电极材料. 尽管非贵过渡金属基催化剂材料价格低廉、储量丰富,在电解水制氢领域中具有应用前景,但其性能与贵金属相比仍存在差距. 结合先进的表征技术和理论计算,深入研究非贵过渡金属基电催化剂催化分解水的机制,可为制备高催化活性的催化剂提供指导. 同时为实现低成本、大规模合成催化剂,制备方法应综合考虑成本、时间、可操作性和可重复性的因素. 电解质中的H+/OH−在长期运行中被不断消耗,导致电极材料表面局部pH变化,从而可能影响材料的催化活性和结构稳定性. 因此,开发酸性/中性电解质中的催化剂材料具有重要意义.
(2)深入研究阳极替代反应以及设计新型的电解槽. 热力学电位更低的氧化反应能够有效替代OER,有效降低制氢的电能消耗. 然而现阶段针对反应机理的研究及催化剂的设计尚在起步阶段. 此外,阳极反应物的成本和氧化产物的经济价值也是影响制氢经济效益的重要因素. 选择合适的阳极替代反应才能实现经济效益的最大化. 阳极反应物可能发生不完全氧化,生成成分复杂的产物,会增加产品的提纯成本,故设计高选择性的催化剂是未来研究的重点. 替代OER的阳极氧化反应类型丰富,其氧化产物也呈现多样性. 利用阳极替代反应辅助制氢面临着反应物/产物腐蚀隔膜以及高附加值产物分离提纯等实际问题,因此需要根据特定的要求设计配置新型的电解槽.
-
![]()
图 1 (a) 电解槽示意图; (b) HER和OER的电极电位随pH变化的Pourbaix图
Figure 1. (a) Scheme of a water electrolyzer; (b) Pourbaix plot of electrode potential vs pH for the HER and OER
![]()
图 2 (a)~(b) OER过程中的AEM机制; (c)~(e) OER过程中的LOM机制[14]. 其中,M代表金属元素,蓝色O代表电解液中的羟基氧,红色O代表晶格氧,方框代表氧空位
Figure 2. (a)–(b) AEM mechanism scheme of the OER; (c)–(e) LOM mechanism scheme of the OER[14]. M represents the metal element, blue O, red O, and box represents the oxygen in hydroxyl, the lattice oxygen, and the box represents the oxygen vacancy, respectively
![]()
图 3 理想条件下和实际应用中,HER(红色实线)和OER(蓝色实线)的线性扫描伏安(Linear sweep voltammetry, LSV)曲线
Figure 3. Linear sweep voltammetry (LSV) curves for the HER (red) and the OER (blue) on practical (dashed) and ideal (solid) electrocatalysts, respectively
![]()
图 4 热力学平衡电位低于OER的阳极替代反应、HER和OER的LSV曲线
Figure 4. LSV curves for anodic substitute reactions, HER, and OER
![]()
图 5 阴离子体相掺杂和表面掺杂的S‒Ni3Se4和Se‒Ni3S2的(a) *H吸附示意图和(b) 活性位点的ΔG*H[22]; (c) 单原子掺杂CoSe2–DETA纳米带的能谱[25]; (d) NiCo2O4和Cr‒NiCo2O4 纳米针的析氢LSV曲线及(e) 纳米针中电化学活性区域和惰性区域示意图[26].
Figure 5. (a) Hydrogen adsorption scheme and (b) ΔG*H for bulk- and surface-doped S‒Ni3Se4 and Se‒Ni3S2[22]; (c) elemental mapping images of CoSe2–DETA nanobelts doped with different metallic single atoms[25]; (d) LSV curves for the HER on pure NiCo2O4 and Cr-doped NiCo2O4; (e) scheme of poorly conductive sites on NiCo2O4 nanoneedles and their activation for the HER from Cr-regulated conductivity[26]
![]()
图 6 (a) Ni3Sn2、Sn掺杂NiO(NiO:Sn)和Ni3Sn2–NiO:Sn吉布斯自由能能垒图,其中TS1表示过渡态1,TS2表示过渡态2;Ni3Sn2-NiSnOx (b)对中间物种的吸附能; (c) 电荷密度分布以及HER机理示意图[29]. (a)~(c) 中蓝色、红色、灰色和天蓝色的小球分别代表Ni, O, Sn和H原子. Ni–NiO/PGC的 (d) 高分辨透射(HRTEM)图; (e) Ni活性位点的HER机理示意图[30]. MoS2/CB的 (f) 结构示意图; (g) LSV曲线和HRTEM图[31]. 在H2O2中刻蚀不同时间的MoS2-x s(x=10~150)(h) MoS2-60 s的STEM图; (i) 电子顺磁共振(EPR)谱; (j) HER的LSV曲线[34]
Figure 6. (a) Energy profiles for the HER on Ni3Sn2, Sn-doped NiO (NiO: Sn) and Ni3Sn2–NiO: Sn; TS1: transition state 1, TS2: transition state 2; (b) adsorption energies of intermediate species and (c) charge density difference for the HER on Ni3Sn2–NiSnOx[29]. Blue, red, gray, and small sky-blue spheres represent Ni, O, Sn, and H atoms, respectively. (d) HRTEM images and (e) HER mechanism on the Ni–NiO heterostructure[30]. (f) Structure of and (g) HER LSV curves for the MoS2/CB[31]. (h) STEM image for MoS2-60 s. (i) EPR spectra and HER LSV curves for MoS2-x etched for different times in H2O2 (x=10–150)[34]
![]()
图 7 α、β、δ、α/β、α/δ和β/δ相的FeOOH (a) XRD图; (b) OER的LSV曲线[44]; (c) Fe2O3–CuO(Fe 位点)的OER过程示意图; (d) 过电位负值与ΔG*O–ΔG*OH之间的火山型关系图[47]; (e) NiFe LDH-VNi和NiFe LDH-VFe催化剂示意图[50]; (f) NixFey(OH)2和 (g) S-NixFey(OH)2表面重构脱氢能垒图; (h) 不同催化剂OER的LSV曲线[57]
Figure 7. (a) XRD patterns and (b) LSV curves for the OER on α, β, δ, α/β, α/δ, and β/δ phases FeOOH[44]; (c) scheme of the OER on the Fe2O3–CuO (Fe site) heterostructure; (d) Overpotentials plotted against a descriptor of ΔG*O–ΔG*OH[47]; (e) Scheme of NiFe LDH-VNi and NiFe LDH-VFe synthesized by alkali etching of the LDHs[50]; Energetic profiles for dehydrogenation on (f) NixFey(OH)2 and (g) S-NixFey(OH)2; (h) LSV curves of OER for different catalysts[57].
![]()
图 8 (a) Fe2O3–NiO/NF催化剂在有无甲醇电解液中的LSV 曲线[66]; (b) 乙醇在NiCuSe、NiFeSe和NiCoSe催化剂表面发生氧化过程所涉及中间产物的吸附能图; (c) 两电极在有无乙醇(ethanol, EtOH)电解液中的LSV曲线[67]
Figure 8. (a) Anodic polarization curves on Fe2O3/NiO-NF with and without the addition of methanol[66]; (b) energetic profile on the NiCoSe, NiFeSe, and NiCoSe; (c) LSV curves obtained on the Pt and NiCoSe catalysts with and without the addition of ethanol[67], ethanol is denoted as EtOH
![]()
图 9 (a) Ni(OH)2@NF氧化尿素的示意图,在有无尿素电解液中的LSV曲线以及稳定性[70]; (b) PW-Co3N NWA/NF催化剂组成的两电极在有无水合肼的电解液中的LSV曲线[73],其中,OHzS表示混合水合肼的水分解,OWS表示全水解; (c) N-NiZnCu LDH/rGO在有无0.3 mol·L−1 NH4Cl的1.0 mol·L−1 KOH电解液中的LSV曲线[74]
Figure 9. (a) Scheme for urea oxidation, together with the LSV curves with and without urea electrolyte, and the chronopotentiometry plot for Ni(OH)2@NF catalyst[70]; (b) LSV curves of PW-Co3N NWA/NF as both the anode and cathode with and without the addition of hydrazine[73]; Overall hydrazine splitting is denoted as OHzS and overall water splitting is denoted as OWS; (c) LSV curves of N-NiZnCu LDH/rGO in 1.0 mol·L−1 KOH with and without 0.3 mol·L−1 NH4Cl[74
![]()
![]()
图 11 非贵过渡金属基催化剂参与HER和OER的反应机理、催化剂性能优化策略、阳极替代反应辅助制氢的策略以及电解水制氢的工程应用前景概述
Figure 11. Overview of HER and OER mechanisms of noble-metal-free catalysts, optimization strategies, substitute oxidation reaction-assisted hydrogen production strategies, and engineering application prospects of hydrogen production by water splitting
-
[1] Jiao Y, Zheng Y, Jaroniec M, et al. Design of electrocatalysts for oxygen- and hydrogen-involving energy conversion reactions. Chem Soc Rev, 2015, 44(8): 2060 doi: 10.1039/C4CS00470A
[2] Turner J A. A realizable renewable energy future. Science, 1999, 285(5428): 687 doi: 10.1126/science.285.5428.687
[3] Andrews J, Shabani B. Re-envisioning the role of hydrogen in a sustainable energy economy. Int J Hydrogen Energy, 2012, 37(2): 1184 doi: 10.1016/j.ijhydene.2011.09.137
[4] Dincer I. Green methods for hydrogen production. Int J Hydrogen Energy, 2012, 37(2): 1954 doi: 10.1016/j.ijhydene.2011.03.173
[5] 国家能源局. 氢能产业发展中长期规划(2021—2035年)[J/OL]. 国家能源局网站 (2022–03–23) [2024–07–20]. https://zfxxgk.nea.gov.cn/2022-03/23/c_1310525630.htm National Energy Administration, Medium and long-term plan for the development of hydrogen energy industry(2021—2035) [J/OL]. National Energy Administration Website (2022–03–23) [2024–07–20]. https://zfxxgk.nea.gov.cn/2022-03/23/c_1310525630.htm
[6] Zhang J, Zhang Q Y, Feng X L. Support and interface effects in water-splitting electrocatalysts. Adv Mater, 2019, 31(31): 1808167 doi: 10.1002/adma.201808167
[7] Zou X X, Zhang Y. Noble metal-free hydrogen evolution catalysts for water splitting. Chem Soc Rev, 2015, 44(15): 5148 doi: 10.1039/C4CS00448E
[8] Zheng Y, Jiao Y, Jaroniec M, et al. Advancing the electrochemistry of the hydrogen-evolution reaction through combining experiment and theory. Angew Chem Int Ed, 2015, 54(1): 52 doi: 10.1002/anie.201407031
[9] Liu F, Shi C X, Guo X L, et al. Rational design of better hydrogen evolution electrocatalysts for water splitting: A review. Adv Sci, 2022, 9(18): 2200307 doi: 10.1002/advs.202200307
[10] Fu X W, Shi R J, Jiao S L, et al. Structural design for electrocatalytic water splitting to realize industrial-scale deployment: Strategies, advances, and perspectives. J Energy Chem, 2022, 70: 129 doi: 10.1016/j.jechem.2022.02.010
[11] Suen N T, Hung S F, Quan Q, et al. Electrocatalysis for the oxygen evolution reaction: Recent development and future perspectives. Chem Soc Rev, 2017, 46(2): 337 doi: 10.1039/C6CS00328A
[12] Fabbri E, Nachtegaal M, Binninger T, et al. Dynamic surface self-reconstruction is the key of highly active perovskite nano-electrocatalysts for water splitting. Nat Mater, 2017, 16(9): 925 doi: 10.1038/nmat4938
[13] Grimaud A, Diaz-Morales O, Han B H, et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat Chem, 2017, 9(5): 457 doi: 10.1038/nchem.2695
[14] Zhou B H, Gao R J, Zou J J, et al. Surface design strategy of catalysts for water electrolysis. Small, 2022, 18(27): 2202336 doi: 10.1002/smll.202202336
[15] Sun H N, Xu X M, Kim H, et al. Electrochemical water splitting: Bridging the gaps between fundamental research and industrial applications. Energy Environ Mater, 2023, 6(5): e12441 doi: 10.1002/eem2.12441
[16] Wang T Z, Cao X J, Jiao L F. Progress in hydrogen production coupled with electrochemical oxidation of small molecules. Angew Chem Int Ed, 2022, 61(51): e202213328 doi: 10.1002/anie.202213328
[17] Veeramani K, Janani G, Kim J, et al. Hydrogen and value-added products yield from hybrid water electrolysis: A critical review on recent developments. Renewable Sustainable Energy Rev, 2023, 177: 113227 doi: 10.1016/j.rser.2023.113227
[18] Ren J T, Chen L, Wang H Y, et al. Water electrolysis for hydrogen production: From hybrid systems to self-powered/catalyzed devices. Energy Environ Sci, 2024, 17(1): 49 doi: 10.1039/D3EE02467A
[19] Sabatier P. La Catalyse en Chimie Organique. Paris: Librairie Polytechnique, Ch. Beranger, 1920.
[20] Nørskov J K, Bligaard T, Logadottir A, et al. Trends in the exchange current for hydrogen evolution. J Electrochem Soc, 2005, 152(3): J23 doi: 10.1149/1.1856988
[21] Xiong L W, Qiu Y F, Peng X, et al. Electronic structural engineering of transition metal-based electrocatalysts for the hydrogen evolution reaction. Nano Energy, 2022, 104: 107882 doi: 10.1016/j.nanoen.2022.107882
[22] Liu T Y, Diao P, Lin Z, et al. Sulfur and selenium doped nickel chalcogenides as efficient and stable electrocatalysts for hydrogen evolution reaction: The importance of the dopant atoms in and beneath the surface. Nano Energy, 2020, 74: 104787 doi: 10.1016/j.nanoen.2020.104787
[23] Xue N, Lin Z, Li P K, et al. Sulfur-doped CoSe2 porous nanosheets as efficient electrocatalysts for the hydrogen evolution reaction. ACS Appl Mater Interfaces, 2020, 12(25): 28288 doi: 10.1021/acsami.0c07088
[24] Zhang X L, Yu P C, Su X Z, et al. Efficient acidic hydrogen evolution in proton exchange membrane electrolyzers over a sulfur-doped marcasite-type electrocatalyst. Sci Adv, 2023, 9(27): eadh2885 doi: 10.1126/sciadv.adh2885
[25] Wu R, Xu J, Zhao C L, et al. Dopant triggered atomic configuration activates water splitting to hydrogen. Nat Commun, 2023, 14(1): 2306 doi: 10.1038/s41467-023-37641-3
[26] Liu T Y, Diao P. Nickel foam supported Cr-doped NiCo2O4/FeOOH nanoneedle arrays as a high-performance bifunctional electrocatalyst for overall water splitting. Nano Res, 2020, 13(12): 3299 doi: 10.1007/s12274-020-3006-3
[27] Lin Z, Li K X, Tong Y, et al. Engineering coupled NiS x–WO2.9 heterostructure as pH-universal electrocatalyst for hydrogen evolution reaction. ChemSusChem, 2023, 16(2): 2201985
[28] Zhang J T, Cheng C Q, Xiao L Y, et al. Construction of Co–Se–W at interfaces of phase-mixed cobalt selenide via spontaneous phase transition for platinum-like hydrogen evolution activity and long-term durability in alkaline and acidic media. Adv Mater, 2024, 36(28): 2401880 doi: 10.1002/adma.202401880
[29] Wang X M, Long G F, Liu B, et al. Rationally modulating the functions of Ni3Sn2–NiSnO x nanocomposite electrocatalysts towards enhanced hydrogen evolution reaction. Angew Chem Int Ed, 2023, 62(19): e202301562 doi: 10.1002/anie.202301562
[30] Li C, Xue J Y, Zhang W, et al. Accelerating water dissociation at carbon supported nanoscale Ni/NiO heterojunction electrocatalysts for high-efficiency alkaline hydrogen evolution. Nano Res, 2023, 16(4): 4742 doi: 10.1007/s12274-022-5194-5
[31] Xue N, Diao P. Composite of few-layered MoS2 grown on carbon black: Tuning the ratio of terminal to total sulfur in MoS2 for hydrogen evolution reaction. J Phys Chem C, 2017, 121(27): 14413 doi: 10.1021/acs.jpcc.7b02522
[32] Xue N, Diao P. Molybdenum diselenide nanolayers prepared on carbon black as an efficient and stable electrocatalyst for hydrogen evolution reaction. J Phys Chem C, 2017, 121(48): 26686 doi: 10.1021/acs.jpcc.7b09590
[33] Song H Q, Li Y H, Shang L, et al. Designed controllable nitrogen-doped carbon-dots-loaded MoP nanoparticles for boosting hydrogen evolution reaction in alkaline medium. Nano Energy, 2020, 72: 104730 doi: 10.1016/j.nanoen.2020.104730
[34] Wang X, Zhang Y W, Si H N, et al. Single-atom vacancy defect to trigger high-efficiency hydrogen evolution of MoS2. J Am Chem Soc, 2020, 142(9): 4298 doi: 10.1021/jacs.9b12113
[35] Jin J, Wang X Y, Hu Y, et al. Precisely control relationship between sulfur vacancy and H absorption for boosting hydrogen evolution reaction. Nano Micro Lett, 2024, 16(1): 63 doi: 10.1007/s40820-023-01291-3
[36] Zhan W Q, Zhai X W, Li Y H, et al. Regulating local atomic environment around vacancies for efficient hydrogen evolution. ACS Nano, 2024, 18(14): 10312 doi: 10.1021/acsnano.4c02283
[37] Zhao G Q, Rui K, Dou S X, et al. Heterostructures for electrochemical hydrogen evolution reaction: A review. Adv Funct Mater, 2018, 28(43): 1803291 doi: 10.1002/adfm.201803291
[38] Man I C, Su H Y, Calle-Vallejo F, et al. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem, 2011, 3(7): 1159 doi: 10.1002/cctc.201000397
[39] Lin Z, Bu P P, Xiao Y, et al. β- and γ-NiFeOOH electrocatalysts for an efficient oxygen evolution reaction: An electrochemical activation energy aspect. J Mater Chem A, 2022, 10(39): 20847 doi: 10.1039/D2TA04688A
[40] Lin Z, Gao Q L, Diao P. Promoting the electrocatalytic oxygen evolution reaction on NiCo2O4 with infrared-thermal effect: A strategy to utilize the infrared solar energy to reduce activation energy during water splitting. J Colloid Interface Sci, 2023, 638: 54 doi: 10.1016/j.jcis.2023.01.130
[41] Lu Z Y, Xu W W, Zhu W, et al. Three-dimensional NiFe layered double hydroxide film for high-efficiency oxygen evolution reaction. Chem Commun, 2014, 50(49): 6479 doi: 10.1039/C4CC01625D
[42] Li S C, Liu T, Zhang W, et al. Highly efficient anion exchange membrane water electrolyzers via chromium-doped amorphous electrocatalysts. Nat Commun, 2024, 15(1): 3416 doi: 10.1038/s41467-024-47736-0
[43] Chong L N, Gao G P, Wen J G, et al. La- and Mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis. Science, 2023, 380(6645): 609 doi: 10.1126/science.ade1499
[44] Hu J, Li S W, Chu J Y, et al. Understanding the phase-induced electrocatalytic oxygen evolution reaction activity on FeOOH nanostructures. ACS Catal, 2019, 9(12): 10705 doi: 10.1021/acscatal.9b03876
[45] He J L, Hu B B, Zhao Y. Superaerophobic electrode with Metal@Metal-oxide Powder catalyst for oxygen evolution reaction. Adv Funct Mater, 2016, 26(33): 5998 doi: 10.1002/adfm.201602116
[46] Zhao P D, Fu S Q, Luo Y C, et al. Deciphering the space charge effect of the CoNiLDH/FeOOH n–n heterojunction for efficient electrocatalytic oxygen evolution. Small, 2023, 19(52): 2305241 doi: 10.1002/smll.202305241
[47] Hu F, Yu D S, Ye M, et al. Lattice-matching formed mesoporous transition metal oxide heterostructures advance water splitting by active Fe—O—Cu bridges. Adv Energy Mater, 2022, 12(19): 2200067 doi: 10.1002/aenm.202200067
[48] Huang J Z, Sheng H Y, Ross R D, et al. Modifying redox properties and local bonding of Co3O4 by CeO2 enhances oxygen evolution catalysis in acid. Nat Commun, 2021, 12(1): 3036 doi: 10.1038/s41467-021-23390-8
[49] Yan D F, Xia C F, Zhang W J, et al. Cation defect engineering of transition metal electrocatalysts for oxygen evolution reaction. Adv Energy Mater, 2022, 12(45): 2202317 doi: 10.1002/aenm.202202317
[50] Wang Y Y, Qiao M, Li Y F, et al. Tuning surface electronic configuration of NiFe LDHs nanosheets by introducing cation vacancies (Fe or Ni) as highly efficient electrocatalysts for oxygen evolution reaction. Small, 2018, 14(17): 1800136 doi: 10.1002/smll.201800136
[51] Xu L, Jiang Q Q, Xiao Z H, et al. Plasma-engraved Co3O4 nanosheets with oxygen vacancies and high surface area for the oxygen evolution reaction. Angew Chem Int Ed, 2016, 55(17): 5277 doi: 10.1002/anie.201600687
[52] Peng L S, Yang N, Yang Y Q, et al. Atomic cation-vacancy engineering of NiFe-layered double hydroxides for improved activity and stability towards the oxygen evolution reaction. Angew Chem Int Ed, 2021, 60(46): 24612 doi: 10.1002/anie.202109938
[53] Wu L, Qin H L, Ji Z Y, et al. Nitrogen-doped carbon dots modified Fe–Co sulfide nanosheets as high-efficiency electrocatalysts toward oxygen evolution reaction. Small, 2024, 20(4): 2305965 doi: 10.1002/smll.202305965
[54] Cui X J, Ren P J, Deng D H, et al. Single layer graphene encapsulating non-precious metals as high-performance electrocatalysts for water oxidation. Energy Environ Sci, 2016, 9(1): 123 doi: 10.1039/C5EE03316K
[55] Zhao C X, Liu J N, Wang J, et al. A clicking confinement strategy to fabricate transition metal single-atom sites for bifunctional oxygen electrocatalysis. Sci Adv, 2022, 8(11): eabn5091 doi: 10.1126/sciadv.abn5091
[56] Li X Y, Niu Z G, Jiang J, et al. Cobalt nanoparticles embedded in porous N-rich carbon as an efficient bifunctional electrocatalyst for water splitting. J Mater Chem A, 2016, 4(9): 3204 doi: 10.1039/C6TA00223D
[57] Lei H, Ma L, Wan Q X, et al. Promoting surface reconstruction of NiFe layered double hydroxide for enhanced oxygen evolution. Adv Energy Mater, 2022, 12(48): 2202522 doi: 10.1002/aenm.202202522
[58] Chen J W, Chen H X, Yu T W, et al. Recent advances in the understanding of the surface reconstruction of oxygen evolution electrocatalysts and materials development. Electrochem Energy Rev, 2021, 4(3): 566 doi: 10.1007/s41918-021-00104-8
[59] Huang Y, Zhang L, Jiang L W, et al. Electronic structure regulation and surface reconstruction of iron diselenide for enhanced oxygen evolution activity. Small, 2023, 19(48): 2302970 doi: 10.1002/smll.202302970
[60] Han L, Dong S J, Wang E K. Transition-metal (co, Ni, and Fe)-based electrocatalysts for the water oxidation reaction. Adv Mater, 2016, 28(42): 9266 doi: 10.1002/adma.201602270
[61] Hu C, Li M Y, Qiu J S, et al. Design and fabrication of carbon dots for energy conversion and storage. Chem Soc Rev, 2019, 48(8): 2315 doi: 10.1039/C8CS00750K
[62] Deng C, Toe C Y, Li X, et al. Earth-abundant metal-based electrocatalysts promoted anodic reaction in hybrid water electrolysis for efficient hydrogen production: Recent progress and perspectives. Adv Energy Mater, 2022, 12(25): 2201047 doi: 10.1002/aenm.202201047
[63] Quan L, Jiang H, Mei G L, et al. Bifunctional electrocatalysts for overall and hybrid water splitting. Chem Rev, 2024, 124(7): 3694 doi: 10.1021/acs.chemrev.3c00332
[64] Qian Q Z, Zhu Y, Ahmad N, et al. Recent advancements in electrochemical hydrogen production via hybrid water splitting. Adv Mater, 2024, 36(4): 2306108 doi: 10.1002/adma.202306108
[65] Zhao B, Liu J W, Yin Y R, et al. Carbon nanofibers@NiSe core/sheath nanostructures as efficient electrocatalysts for integrating highly selective methanol conversion and less-energy intensive hydrogen production. J Mater Chem A, 2019, 7(45): 25878 doi: 10.1039/C9TA09782A
[66] Hao Y X, Yu D S, Zhu S Q, et al. Methanol upgrading coupled with hydrogen product at large current density promoted by strong interfacial interactions. Energy Environ Sci, 2023, 16(3): 1100 doi: 10.1039/D2EE03936B
[67] Li X L, Chen M C, Ye Y T, et al. Electronic structure modulation of nickel sites by cationic heterostructures to optimize ethanol electrooxidation activity in alkaline solution. Small, 2023, 19(18): e2207086 doi: 10.1002/smll.202207086
[68] Xia Z C, Ma C Y, Fan Y, et al. Vacancy optimized coordination on nickel oxide for selective electrocatalytic oxidation of glycerol. ACS Catal, 2024, 14(3): 1930 doi: 10.1021/acscatal.3c04568
[69] Xu S, Ruan X W, Ganesan M, et al. Transition metal-based catalysts for urea oxidation reaction (UOR): Catalyst design strategies, applications, and future perspectives. Adv Funct Mater, 2024, 34(18): 2313309 doi: 10.1002/adfm.202313309
[70] Xia L Y, Liao Y, Qing Y, et al. In situ growth of porous ultrathin Ni(OH)2 nanostructures on nickel foam: An efficient and durable catalysts for urea electrolysis. ACS Appl Energy Mater, 2020, 3(3): 2996 doi: 10.1021/acsaem.0c00122
[71] Du M X, Ji Y J, Li Y Y, et al. Construction of an internal charge field: CoS1.097/Ni3S2 heterojunction promotes efficient urea oxidation reaction. Adv Funct Materials, 2024, 34(38): 2402776 doi: 10.1002/adfm.202402776
[72] Tang C, Zhang R, Lu W B, et al. Energy-saving electrolytic hydrogen generation: Ni2 P nanoarray as a high-performance non-noble-metal electrocatalyst. Angew Chem Int Ed, 2017, 56(3): 842 doi: 10.1002/anie.201608899
[73] Liu Y, Zhang J H, Li Y P, et al. Manipulating dehydrogenation kinetics through dual-doping Co3N electrode enables highly efficient hydrazine oxidation assisting self-powered H2 production. Nat Commun, 2020, 11(1): 1853 doi: 10.1038/s41467-020-15563-8
[74] Hu S N, Tan Y, Feng C Q, et al. Synthesis of N doped NiZnCu-layered double hydroxides with reduced graphene oxide on nickel foam as versatile electrocatalysts for hydrogen production in hybrid-water electrolysis. J Power Sources, 2020, 453: 227872 doi: 10.1016/j.jpowsour.2020.227872
[75] Chen F Y, Wu Z Y, Adler Z, et al. Stability challenges of electrocatalytic oxygen evolution reaction: From mechanistic understanding to reactor design. Joule, 2021, 5(7): 1704 doi: 10.1016/j.joule.2021.05.005
[76] Chatenet M, Pollet B G, Dekel D R, et al. Water electrolysis: From textbook knowledge to the latest scientific strategies and industrial developments. Chem Soc Rev, 2022, 51(11): 4583 doi: 10.1039/D0CS01079K
[77] Hall D E. Electrodes for alkaline water electrolysis. J Electrochem Soc, 1981, 128(4): 740 doi: 10.1149/1.2127498
[78] Shiva Kumar S, Himabindu V. Hydrogen production by PEM water electrolysis–A review. Mater Sci Energy Technol, 2019, 2(3): 442
[79] Wu X, Scott K. Cu xCo3− xO4 (0≤x<1) nanoparticles for oxygen evolution in high performance alkaline exchange membranewater electrolysers. J Mater Chem, 2011, 21(33): 12344 doi: 10.1039/c1jm11312g
[80] Vincent I, Bessarabov D. Low cost hydrogen production by anion exchange membrane electrolysis: A review. Renewable Sustainable Energy Rev, 2018, 81: 1690 doi: 10.1016/j.rser.2017.05.258
[81] 李海鹏, 孙邦兴, 李嘉烨. 双碳目标下绿色制氢技术的进展. 电池, 2024, 54(2):271 Li H P, Sun B X, Li J Y. Progress of green hydrogen production technology under dual carbon goal. Battery Bimonthly, 2024, 54(2): 271
[82] Feng C R, Chen M, Yang Z Y, et al. Electrocatalytic seawater splitting for hydrogen production: Recent progress and future prospects. J Mater Sci Technol, 2023, 162: 203 doi: 10.1016/j.jmst.2023.03.058
[83] Jin H Y, Xu J, Liu H, et al. Emerging materials and technologies for electrocatalytic seawater splitting. Sci Adv, 2023, 9(42): eadi7755 doi: 10.1126/sciadv.adi7755
[84] Chen L, Yu C, Dong J T, et al. Seawater electrolysis for fuels and chemicals production: Fundamentals, achievements, and perspectives. Chem Soc Rev, 2024, 53(14): 7455 doi: 10.1039/D3CS00822C
[85] Xie H P, Zhao Z Y, Liu T, et al. A membrane-based seawater electrolyser for hydrogen generation. Nature, 2022, 612(7941): 673 doi: 10.1038/s41586-022-05379-5
[86] Na J C, Yu H M, Jia S Y, et al. Electrochemical reconstruction of non-noble metal-based heterostructure nanorod arrays electrodes for highly stable anion exchange membrane seawater electrolysis. J Energy Chem, 2024, 91: 370 doi: 10.1016/j.jechem.2023.12.018
[87] 王淼, 邓蓉蓉, 张启波. 电解水阳极析氧替代反应及高效催化剂研究进展. 工程科学学报, 2024, 46(4):744 Wang M, Deng R R, Zhang Q B. Recent advances in alternative oxidation reactions for water splitting and their efficient electrocatalysts. Chin J Eng, 2024, 46(4): 744
[88] 张唯怡, 张议洁, 王进伟, 等. 电解水制氢技术及大电流析氧反应研究与展望. 工程科学学报, 2023, 45(7):1057 Zhang W Y, Zhang Y J, Wang J W, et al. Research and perspectives on electrocatalytic water splitting and large current density oxygen evolution reaction. Chin J Eng, 2023, 45(7): 1057
[89] Wang H Y, Sun M L, Ren J T, et al. Circumventing challenges: Design of anodic electrocatalysts for hybrid water electrolysis systems. Adv Energy Mater, 2023, 13(4): 2203568 doi: 10.1002/aenm.202203568
 下载:
下载:
计量
- 文章访问数: 349
- HTML全文浏览量: 269
- PDF下载量: 53
